Sparse pseudocontact shift NMR data obtained from a non-canonical amino acid-linked lanthanide tag improves integral membrane protein structure prediction
Kaitlyn V. Ledwitch, Georg Künze, Jacob R. McKinney, Elleansar Okwei, Katherine Larochelle, Lisa Pankewitz, Soumya Ganguly, Heather L. Darling, Irene Coin, Jens Meiler
{"title":"Sparse pseudocontact shift NMR data obtained from a non-canonical amino acid-linked lanthanide tag improves integral membrane protein structure prediction","authors":"Kaitlyn V. Ledwitch, Georg Künze, Jacob R. McKinney, Elleansar Okwei, Katherine Larochelle, Lisa Pankewitz, Soumya Ganguly, Heather L. Darling, Irene Coin, Jens Meiler","doi":"10.1007/s10858-023-00412-9","DOIUrl":null,"url":null,"abstract":"<div><p>A single experimental method alone often fails to provide the resolution, accuracy, and coverage needed to model integral membrane proteins (IMPs). Integrating computation with experimental data is a powerful approach to supplement missing structural information with atomic detail. We combine RosettaNMR with experimentally-derived paramagnetic NMR restraints to guide membrane protein structure prediction. We demonstrate this approach using the disulfide bond formation protein B (DsbB), an α-helical IMP. Here, we attached a cyclen-based paramagnetic lanthanide tag to an engineered non-canonical amino acid (ncAA) using a copper-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry reaction. Using this tagging strategy, we collected 203 backbone H<sup>N</sup> pseudocontact shifts (PCSs) for three different labeling sites and used these as input to guide <i>de novo</i> membrane protein structure prediction protocols in Rosetta. We find that this sparse PCS dataset combined with 44 long-range NOEs as restraints in our calculations improves structure prediction of DsbB by enhancements in model accuracy, sampling, and scoring. The inclusion of this PCS dataset improved the Cα-RMSD transmembrane segment values of the best-scoring and best-RMSD models from 9.57 Å and 3.06 Å (no NMR data) to 5.73 Å and 2.18 Å, respectively.</p></div>","PeriodicalId":613,"journal":{"name":"Journal of Biomolecular NMR","volume":"77 3","pages":"69 - 82"},"PeriodicalIF":1.9000,"publicationDate":"2023-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10858-023-00412-9.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Biomolecular NMR","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10858-023-00412-9","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
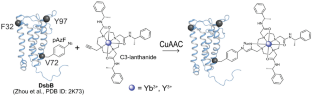
A single experimental method alone often fails to provide the resolution, accuracy, and coverage needed to model integral membrane proteins (IMPs). Integrating computation with experimental data is a powerful approach to supplement missing structural information with atomic detail. We combine RosettaNMR with experimentally-derived paramagnetic NMR restraints to guide membrane protein structure prediction. We demonstrate this approach using the disulfide bond formation protein B (DsbB), an α-helical IMP. Here, we attached a cyclen-based paramagnetic lanthanide tag to an engineered non-canonical amino acid (ncAA) using a copper-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry reaction. Using this tagging strategy, we collected 203 backbone HN pseudocontact shifts (PCSs) for three different labeling sites and used these as input to guide de novo membrane protein structure prediction protocols in Rosetta. We find that this sparse PCS dataset combined with 44 long-range NOEs as restraints in our calculations improves structure prediction of DsbB by enhancements in model accuracy, sampling, and scoring. The inclusion of this PCS dataset improved the Cα-RMSD transmembrane segment values of the best-scoring and best-RMSD models from 9.57 Å and 3.06 Å (no NMR data) to 5.73 Å and 2.18 Å, respectively.
期刊介绍:
The Journal of Biomolecular NMR provides a forum for publishing research on technical developments and innovative applications of nuclear magnetic resonance spectroscopy for the study of structure and dynamic properties of biopolymers in solution, liquid crystals, solids and mixed environments, e.g., attached to membranes. This may include:
Three-dimensional structure determination of biological macromolecules (polypeptides/proteins, DNA, RNA, oligosaccharides) by NMR.
New NMR techniques for studies of biological macromolecules.
Novel approaches to computer-aided automated analysis of multidimensional NMR spectra.
Computational methods for the structural interpretation of NMR data, including structure refinement.
Comparisons of structures determined by NMR with those obtained by other methods, e.g. by diffraction techniques with protein single crystals.
New techniques of sample preparation for NMR experiments (biosynthetic and chemical methods for isotope labeling, preparation of nutrients for biosynthetic isotope labeling, etc.). An NMR characterization of the products must be included.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
