Maxime Lepetit, Mirela Diana Ilie, Marie Chanal, Gerald Raverot, Philippe Bertolino, Christophe Arpin, Franck Picard, Olivier Gandrillon
{"title":"scAN1.0: A reproducible and standardized pipeline for processing 10X single cell RNAseq data.","authors":"Maxime Lepetit, Mirela Diana Ilie, Marie Chanal, Gerald Raverot, Philippe Bertolino, Christophe Arpin, Franck Picard, Olivier Gandrillon","doi":"10.3233/ISB-220252","DOIUrl":null,"url":null,"abstract":"<p><p>Single cell transcriptomics has recently seen a surge in popularity, leading to the need for data analysis pipelines that are reproducible, modular, and interoperable across different systems and institutions.To meet this demand, we introduce scAN1.0, a processing pipeline for analyzing 10X single cell RNA sequencing data. scAN1.0 is built using the Nextflow DSL2 and can be run on most computational systems. The modular design of Nextflow pipelines enables easy integration and evaluation of different blocks for specific analysis steps.We demonstrate the usefulness of scAN1.0 by showing its ability to examine the impact of the mapping step during the analysis of two datasets: (i) a 10X scRNAseq of a human pituitary gonadotroph tumor dataset and (ii) a murine 10X scRNAseq acquired on CD8 T cells during an immune response.</p>","PeriodicalId":39379,"journal":{"name":"In Silico Biology","volume":" ","pages":"11-21"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10741331/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"In Silico Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3233/ISB-220252","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
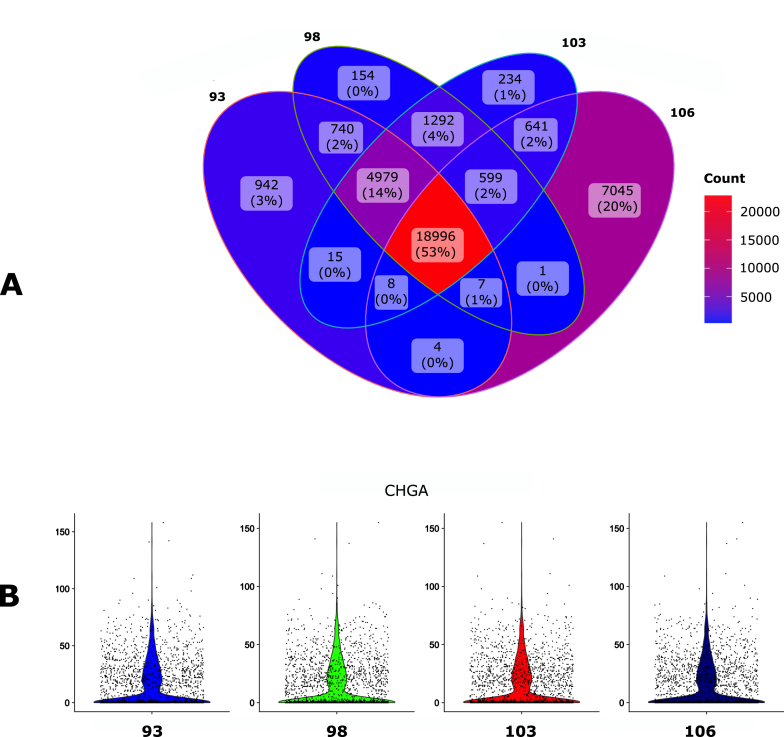
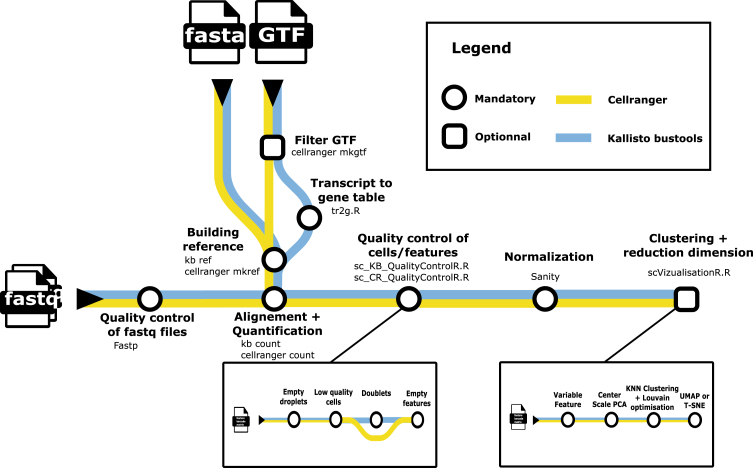
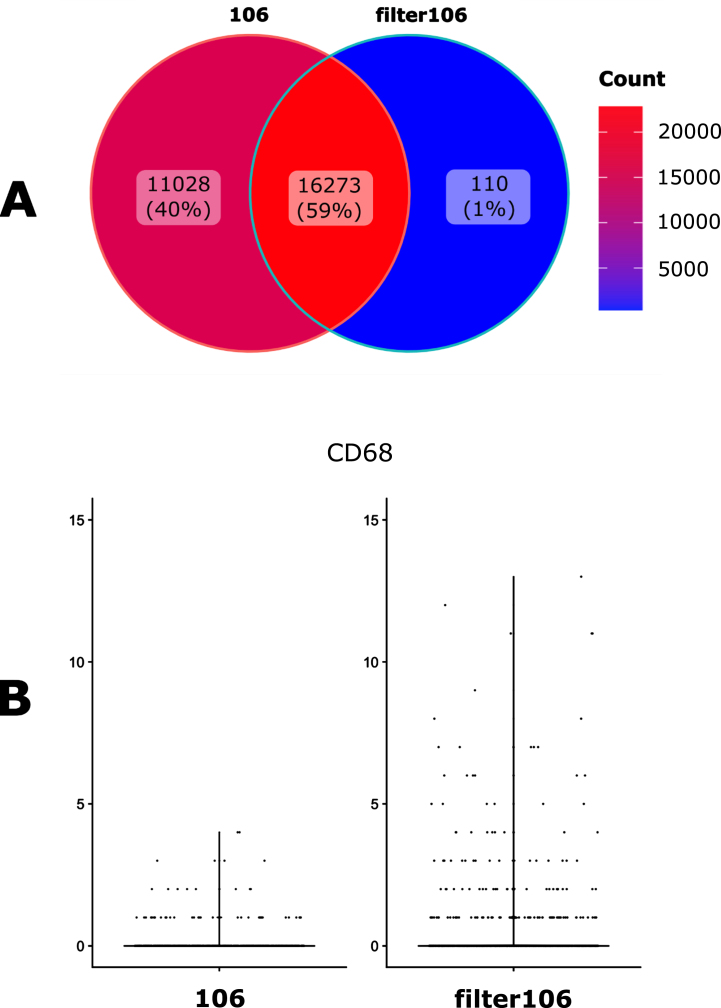
Single cell transcriptomics has recently seen a surge in popularity, leading to the need for data analysis pipelines that are reproducible, modular, and interoperable across different systems and institutions.To meet this demand, we introduce scAN1.0, a processing pipeline for analyzing 10X single cell RNA sequencing data. scAN1.0 is built using the Nextflow DSL2 and can be run on most computational systems. The modular design of Nextflow pipelines enables easy integration and evaluation of different blocks for specific analysis steps.We demonstrate the usefulness of scAN1.0 by showing its ability to examine the impact of the mapping step during the analysis of two datasets: (i) a 10X scRNAseq of a human pituitary gonadotroph tumor dataset and (ii) a murine 10X scRNAseq acquired on CD8 T cells during an immune response.
In Silico BiologyComputer Science-Computational Theory and Mathematics
CiteScore
2.20
自引率
0.00%
发文量
1
期刊介绍:
The considerable "algorithmic complexity" of biological systems requires a huge amount of detailed information for their complete description. Although far from being complete, the overwhelming quantity of small pieces of information gathered for all kind of biological systems at the molecular and cellular level requires computational tools to be adequately stored and interpreted. Interpretation of data means to abstract them as much as allowed to provide a systematic, an integrative view of biology. Most of the presently available scientific journals focus either on accumulating more data from elaborate experimental approaches, or on presenting new algorithms for the interpretation of these data. Both approaches are meritorious.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
