Wubing Zhang , Shourya S. Roy Burman , Jiaye Chen , Katherine A. Donovan , Yang Cao , Chelsea Shu , Boning Zhang , Zexian Zeng , Shengqing Gu , Yi Zhang , Dian Li , Eric S. Fischer , Collin Tokheim , X. Shirley Liu
{"title":"Machine Learning Modeling of Protein-intrinsic Features Predicts Tractability of Targeted Protein Degradation","authors":"Wubing Zhang , Shourya S. Roy Burman , Jiaye Chen , Katherine A. Donovan , Yang Cao , Chelsea Shu , Boning Zhang , Zexian Zeng , Shengqing Gu , Yi Zhang , Dian Li , Eric S. Fischer , Collin Tokheim , X. Shirley Liu","doi":"10.1016/j.gpb.2022.11.008","DOIUrl":null,"url":null,"abstract":"<div><p><strong>Targeted protein degradation</strong> (TPD) has rapidly emerged as a therapeutic modality to eliminate previously undruggable proteins by repurposing the cell’s endogenous protein degradation machinery. However, the susceptibility of proteins for targeting by TPD approaches, termed “<strong>degradability</strong>”, is largely unknown. Here, we developed a <strong>machine learning</strong> model, model-free analysis of protein degradability (MAPD), to predict degradability from features intrinsic to protein targets. MAPD shows accurate performance in predicting kinases that are degradable by TPD compounds [with an area under the precision–recall curve (AUPRC) of 0.759 and an area under the receiver operating characteristic curve (AUROC) of 0.775] and is likely generalizable to independent non-kinase proteins. We found five features with statistical significance to achieve optimal prediction, with <strong>ubiquitination</strong> potential being the most predictive. By structural modeling, we found that E2-accessible ubiquitination sites, but not lysine residues in general, are particularly associated with kinase degradability. Finally, we extended MAPD predictions to the entire proteome to find 964 disease-causing proteins (including proteins encoded by 278 cancer genes) that may be tractable to TPD drug development.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"20 5","pages":"Pages 882-898"},"PeriodicalIF":7.9000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/cc/c8/main.PMC10025769.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1672022922001498","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
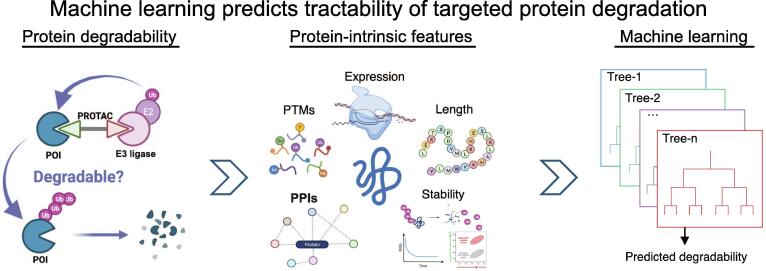
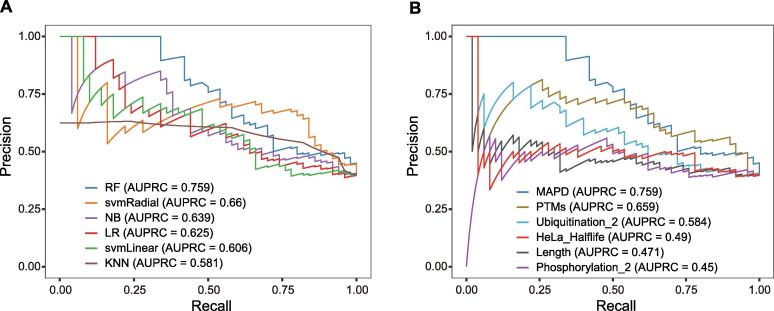
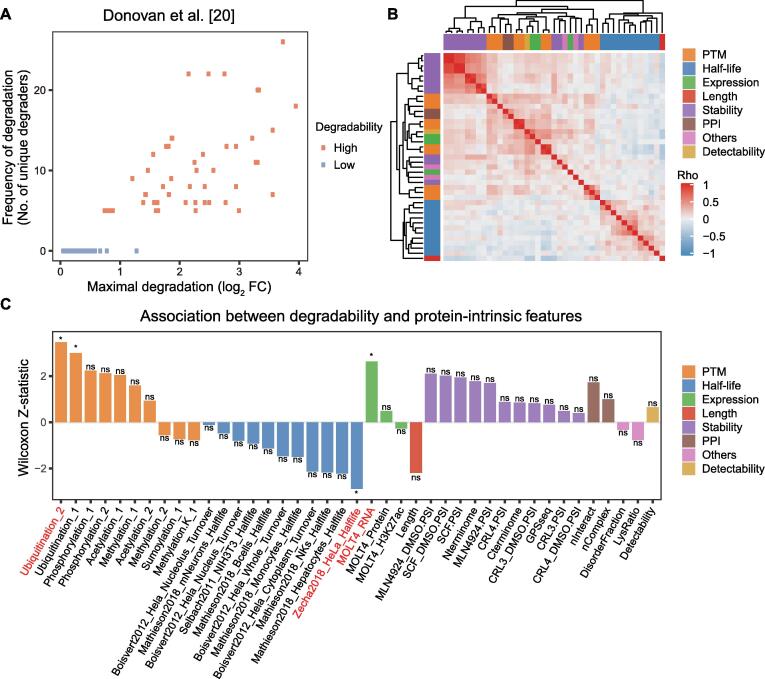
Targeted protein degradation (TPD) has rapidly emerged as a therapeutic modality to eliminate previously undruggable proteins by repurposing the cell’s endogenous protein degradation machinery. However, the susceptibility of proteins for targeting by TPD approaches, termed “degradability”, is largely unknown. Here, we developed a machine learning model, model-free analysis of protein degradability (MAPD), to predict degradability from features intrinsic to protein targets. MAPD shows accurate performance in predicting kinases that are degradable by TPD compounds [with an area under the precision–recall curve (AUPRC) of 0.759 and an area under the receiver operating characteristic curve (AUROC) of 0.775] and is likely generalizable to independent non-kinase proteins. We found five features with statistical significance to achieve optimal prediction, with ubiquitination potential being the most predictive. By structural modeling, we found that E2-accessible ubiquitination sites, but not lysine residues in general, are particularly associated with kinase degradability. Finally, we extended MAPD predictions to the entire proteome to find 964 disease-causing proteins (including proteins encoded by 278 cancer genes) that may be tractable to TPD drug development.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
