Detlef Walter Maria Hofmann, Liudmila Nikolaevna Kuleshova
{"title":"A general force field by machine learning on experimental crystal structures. Calculations of intermolecular Gibbs energy with FlexCryst.","authors":"Detlef Walter Maria Hofmann, Liudmila Nikolaevna Kuleshova","doi":"10.1107/S2053273323000268","DOIUrl":null,"url":null,"abstract":"<p><p>Machine learning was employed on the experimental crystal structures of the Cambridge Structural Database (CSD) to derive an intermolecular force field for all available types of atoms (general force field). The obtained pairwise interatomic potentials of the general force field allow for the fast and accurate calculation of intermolecular Gibbs energy. The approach is based on three postulates regarding Gibbs energy: the lattice energy must be below zero, the crystal structure must be a local minimum, and, if available, the experimental and the calculated lattice energy must coincide. The parametrized general force field was then validated regarding these three conditions. First, the experimental lattice energy was compared with the calculated energies. The observed errors were found to be in the order of experimental errors. Second, Gibbs lattice energy was calculated for all structures available in the CSD. Their energy values were found to be below zero in 99.86% of the cases. Finally, 500 random structures were minimized, and the change in density and energy was examined. The mean error in the case of density was below 4.06%, and for energy it was below 5.7%. The obtained general force field calculated Gibbs lattice energies of 259 041 known crystal structures within a few hours. Since Gibbs energy defines the reaction energy, the calculated energy can be used to predict chemical-physical properties of crystals, for instance, the formation of co-crystals, polymorph stability and solubility.</p>","PeriodicalId":106,"journal":{"name":"Acta Crystallographica Section A: Foundations and Advances","volume":"79 Pt 2","pages":"132-144"},"PeriodicalIF":1.8000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica Section A: Foundations and Advances","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1107/S2053273323000268","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
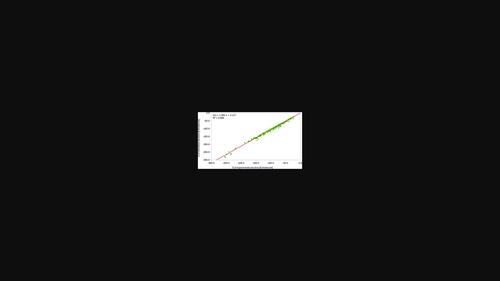
Machine learning was employed on the experimental crystal structures of the Cambridge Structural Database (CSD) to derive an intermolecular force field for all available types of atoms (general force field). The obtained pairwise interatomic potentials of the general force field allow for the fast and accurate calculation of intermolecular Gibbs energy. The approach is based on three postulates regarding Gibbs energy: the lattice energy must be below zero, the crystal structure must be a local minimum, and, if available, the experimental and the calculated lattice energy must coincide. The parametrized general force field was then validated regarding these three conditions. First, the experimental lattice energy was compared with the calculated energies. The observed errors were found to be in the order of experimental errors. Second, Gibbs lattice energy was calculated for all structures available in the CSD. Their energy values were found to be below zero in 99.86% of the cases. Finally, 500 random structures were minimized, and the change in density and energy was examined. The mean error in the case of density was below 4.06%, and for energy it was below 5.7%. The obtained general force field calculated Gibbs lattice energies of 259 041 known crystal structures within a few hours. Since Gibbs energy defines the reaction energy, the calculated energy can be used to predict chemical-physical properties of crystals, for instance, the formation of co-crystals, polymorph stability and solubility.
期刊介绍:
Acta Crystallographica Section A: Foundations and Advances publishes articles reporting advances in the theory and practice of all areas of crystallography in the broadest sense. As well as traditional crystallography, this includes nanocrystals, metacrystals, amorphous materials, quasicrystals, synchrotron and XFEL studies, coherent scattering, diffraction imaging, time-resolved studies and the structure of strain and defects in materials.
The journal has two parts, a rapid-publication Advances section and the traditional Foundations section. Articles for the Advances section are of particularly high value and impact. They receive expedited treatment and may be highlighted by an accompanying scientific commentary article and a press release. Further details are given in the November 2013 Editorial.
The central themes of the journal are, on the one hand, experimental and theoretical studies of the properties and arrangements of atoms, ions and molecules in condensed matter, periodic, quasiperiodic or amorphous, ideal or real, and, on the other, the theoretical and experimental aspects of the various methods to determine these properties and arrangements.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
