{"title":"Atg1, a key regulator of autophagy, functions to promote MAPK activation and cell death upon calcium overload in fission yeast.","authors":"Teruaki Takasaki, Ryosuke Utsumi, Erika Shimada, Asuka Bamba, Kanako Hagihara, Ryosuke Satoh, Reiko Sugiura","doi":"10.15698/mic2023.06.798","DOIUrl":null,"url":null,"abstract":"<p><p>Autophagy promotes or inhibits cell death depending on the environment and cell type. Our previous findings suggested that Atg1 is genetically involved in the regulation of Pmk1 MAPK in fission yeast. Here, we showed that Δ<i>atg1</i> displays lower levels of Pmk1 MAPK phosphorylation than did the wild-type (WT) cells upon treatment with a 1,3-β-D-glucan synthase inhibitor micafungin or CaCl<sub>2</sub>, both of which activate Pmk1. Moreover, the overproduction of Atg1, but not that of the kinase inactivating Atg1<sup>D193A</sup> activates Pmk1 without any extracellular stimuli, suggesting that Atg1 may promote Pmk1 MAPK signaling activation. Notably, the overproduction of Atg1 induces a toxic effect on the growth of WT cells and the deletion of Pmk1 failed to suppress the cell death induced by Atg1, indicating that the Atg1-mediated cell death requires additional mechanism(s) other than Pmk1 activation. Moreover, <i>atg1</i> gene deletion induces tolerance to micafungin and CaCl<sub>2</sub>, whereas <i>pmk1</i> deletion induces severe sensitivities to these compounds. The Δ<i>atg1</i>Δ<i>pmk1</i> double mutants display intermediate sensitivities to these compounds, showing that <i>atg1</i> deletion partly suppressed growth inhibition induced by Δ<i>pmk1</i>. Thus, Atg1 may act to promote cell death upon micafungin and CaCl<sub>2</sub> stimuli regardless of Pmk1 MAPK activity. Since micafungin and CaCl<sub>2</sub> are intracellular calcium inducers, our data reveal a novel role of the autophagy regulator Atg1 to induce cell death upon calcium overload independent of its role in Pmk1 MAPK activation.</p>","PeriodicalId":74183,"journal":{"name":"","volume":"10 6","pages":"133-140"},"PeriodicalIF":0.0,"publicationDate":"2023-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10236205/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.15698/mic2023.06.798","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
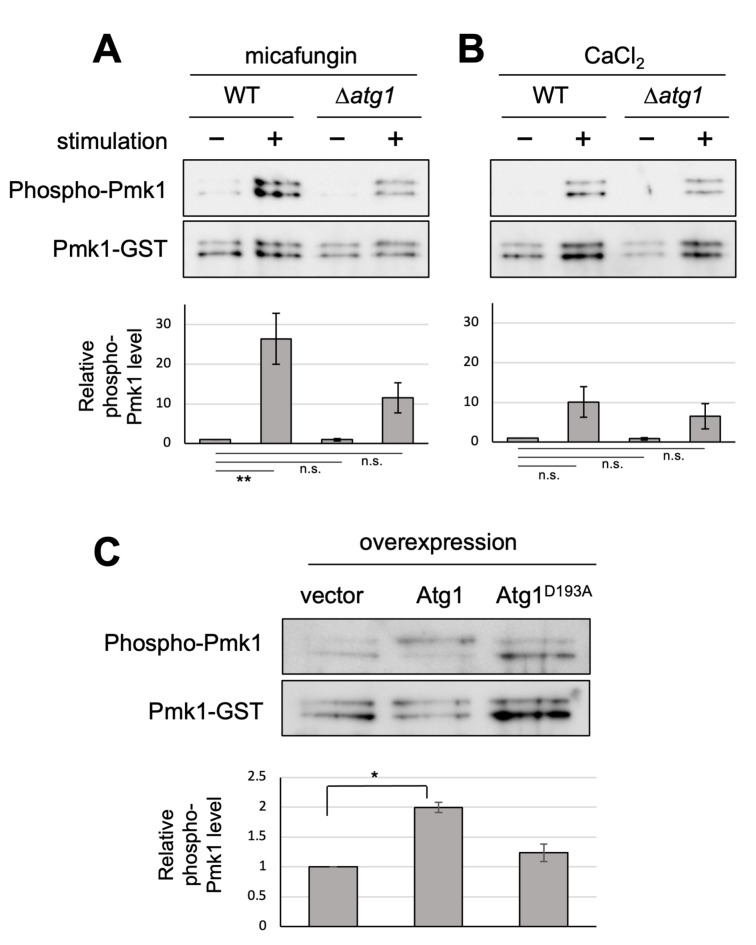
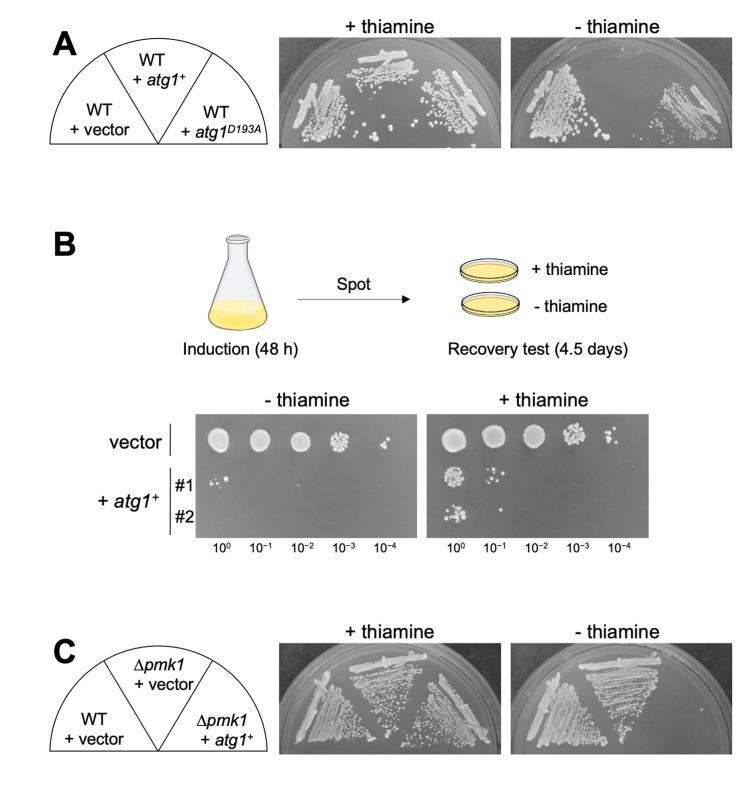
Autophagy promotes or inhibits cell death depending on the environment and cell type. Our previous findings suggested that Atg1 is genetically involved in the regulation of Pmk1 MAPK in fission yeast. Here, we showed that Δatg1 displays lower levels of Pmk1 MAPK phosphorylation than did the wild-type (WT) cells upon treatment with a 1,3-β-D-glucan synthase inhibitor micafungin or CaCl2, both of which activate Pmk1. Moreover, the overproduction of Atg1, but not that of the kinase inactivating Atg1D193A activates Pmk1 without any extracellular stimuli, suggesting that Atg1 may promote Pmk1 MAPK signaling activation. Notably, the overproduction of Atg1 induces a toxic effect on the growth of WT cells and the deletion of Pmk1 failed to suppress the cell death induced by Atg1, indicating that the Atg1-mediated cell death requires additional mechanism(s) other than Pmk1 activation. Moreover, atg1 gene deletion induces tolerance to micafungin and CaCl2, whereas pmk1 deletion induces severe sensitivities to these compounds. The Δatg1Δpmk1 double mutants display intermediate sensitivities to these compounds, showing that atg1 deletion partly suppressed growth inhibition induced by Δpmk1. Thus, Atg1 may act to promote cell death upon micafungin and CaCl2 stimuli regardless of Pmk1 MAPK activity. Since micafungin and CaCl2 are intracellular calcium inducers, our data reveal a novel role of the autophagy regulator Atg1 to induce cell death upon calcium overload independent of its role in Pmk1 MAPK activation.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
