ScInfoVAE: interpretable dimensional reduction of single cell transcription data with variational autoencoders and extended mutual information regularization.
{"title":"ScInfoVAE: interpretable dimensional reduction of single cell transcription data with variational autoencoders and extended mutual information regularization.","authors":"Weiquan Pan, Faning Long, Jian Pan","doi":"10.1186/s13040-023-00333-1","DOIUrl":null,"url":null,"abstract":"<p><p>Single-cell RNA-sequencing (scRNA-seq) data can serve as a good indicator of cell-to-cell heterogeneity and can aid in the study of cell growth by identifying cell types. Recently, advances in Variational Autoencoder (VAE) have demonstrated their ability to learn robust feature representations for scRNA-seq. However, it has been observed that VAEs tend to ignore the latent variables when combined with a decoding distribution that is too flexible. In this paper, we introduce ScInfoVAE, a dimensional reduction method based on the mutual information variational autoencoder (InfoVAE), which can more effectively identify various cell types in scRNA-seq data of complex tissues. A joint InfoVAE deep model and zero-inflated negative binomial distributed model design based on ScInfoVAE reconstructs the objective function to noise scRNA-seq data and learn an efficient low-dimensional representation of it. We use ScInfoVAE to analyze the clustering performance of 15 real scRNA-seq datasets and demonstrate that our method provides high clustering performance. In addition, we use simulated data to investigate the interpretability of feature extraction, and visualization results show that the low-dimensional representation learned by ScInfoVAE retains local and global neighborhood structure data well. In addition, our model can significantly improve the quality of the variational posterior.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"17"},"PeriodicalIF":6.1000,"publicationDate":"2023-06-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10257850/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00333-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
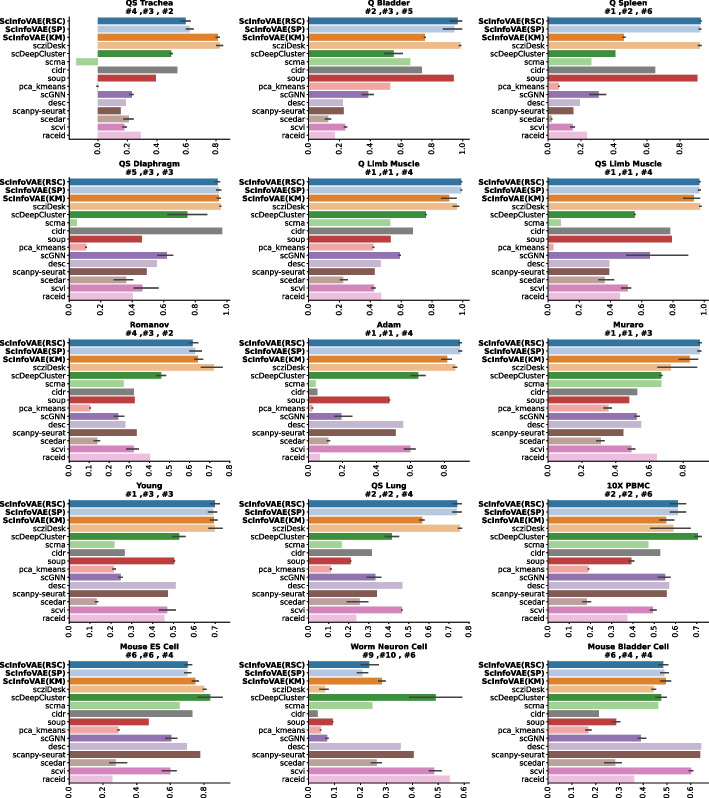
Single-cell RNA-sequencing (scRNA-seq) data can serve as a good indicator of cell-to-cell heterogeneity and can aid in the study of cell growth by identifying cell types. Recently, advances in Variational Autoencoder (VAE) have demonstrated their ability to learn robust feature representations for scRNA-seq. However, it has been observed that VAEs tend to ignore the latent variables when combined with a decoding distribution that is too flexible. In this paper, we introduce ScInfoVAE, a dimensional reduction method based on the mutual information variational autoencoder (InfoVAE), which can more effectively identify various cell types in scRNA-seq data of complex tissues. A joint InfoVAE deep model and zero-inflated negative binomial distributed model design based on ScInfoVAE reconstructs the objective function to noise scRNA-seq data and learn an efficient low-dimensional representation of it. We use ScInfoVAE to analyze the clustering performance of 15 real scRNA-seq datasets and demonstrate that our method provides high clustering performance. In addition, we use simulated data to investigate the interpretability of feature extraction, and visualization results show that the low-dimensional representation learned by ScInfoVAE retains local and global neighborhood structure data well. In addition, our model can significantly improve the quality of the variational posterior.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
