Zinia D'Souza, Irina Pokrovskaya, Vladimir V Lupashin
{"title":"Syntaxin-5's flexibility in SNARE pairing supports Golgi functions.","authors":"Zinia D'Souza, Irina Pokrovskaya, Vladimir V Lupashin","doi":"10.1111/tra.12903","DOIUrl":null,"url":null,"abstract":"<p><p>Deficiency in the conserved oligomeric Golgi (COG) complex that orchestrates SNARE-mediated tethering/fusion of vesicles that recycle the Golgi's glycosylation machinery results in severe glycosylation defects. Although two major Golgi v-SNAREs, GS28/GOSR1, and GS15/BET1L, are depleted in COG-deficient cells, the complete knockout of GS28 and GS15 only modestly affects Golgi glycosylation, indicating the existence of an adaptation mechanism in Golgi SNARE. Indeed, quantitative mass-spectrometry analysis of STX5-interacting proteins revealed two novel Golgi SNARE complexes-STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6. These complexes are present in wild-type cells, but their usage is significantly increased in both GS28- and COG-deficient cells. Upon GS28 deletion, SNAP29 increased its Golgi residency in a STX5-dependent manner. While STX5 depletion and Retro2-induced diversion from the Golgi severely affect protein glycosylation, GS28/SNAP29 and GS28/VTI1B double knockouts alter glycosylation similarly to GS28 KO, indicating that a single STX5-based SNARE complex is sufficient to support Golgi glycosylation. Importantly, co-depletion of three Golgi SNARE complexes in GS28/SNAP29/VTI1B TKO cells resulted in severe glycosylation defects and a reduced capacity for glycosylation enzyme retention at the Golgi. This study demonstrates the remarkable plasticity in SXT5-mediated membrane trafficking, uncovering a novel adaptive response to the failure of canonical intra-Golgi vesicle tethering/fusion machinery.</p>","PeriodicalId":23207,"journal":{"name":"Traffic","volume":"24 8","pages":"355-379"},"PeriodicalIF":2.5000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10330844/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Traffic","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1111/tra.12903","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/21 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
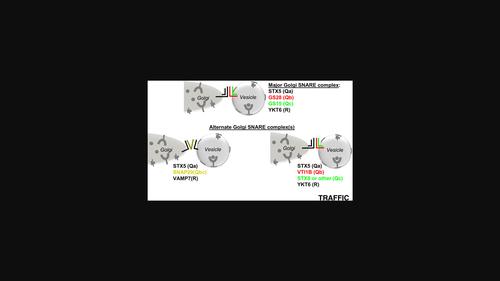
Deficiency in the conserved oligomeric Golgi (COG) complex that orchestrates SNARE-mediated tethering/fusion of vesicles that recycle the Golgi's glycosylation machinery results in severe glycosylation defects. Although two major Golgi v-SNAREs, GS28/GOSR1, and GS15/BET1L, are depleted in COG-deficient cells, the complete knockout of GS28 and GS15 only modestly affects Golgi glycosylation, indicating the existence of an adaptation mechanism in Golgi SNARE. Indeed, quantitative mass-spectrometry analysis of STX5-interacting proteins revealed two novel Golgi SNARE complexes-STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6. These complexes are present in wild-type cells, but their usage is significantly increased in both GS28- and COG-deficient cells. Upon GS28 deletion, SNAP29 increased its Golgi residency in a STX5-dependent manner. While STX5 depletion and Retro2-induced diversion from the Golgi severely affect protein glycosylation, GS28/SNAP29 and GS28/VTI1B double knockouts alter glycosylation similarly to GS28 KO, indicating that a single STX5-based SNARE complex is sufficient to support Golgi glycosylation. Importantly, co-depletion of three Golgi SNARE complexes in GS28/SNAP29/VTI1B TKO cells resulted in severe glycosylation defects and a reduced capacity for glycosylation enzyme retention at the Golgi. This study demonstrates the remarkable plasticity in SXT5-mediated membrane trafficking, uncovering a novel adaptive response to the failure of canonical intra-Golgi vesicle tethering/fusion machinery.
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
