{"title":"A Survey on Methods for Predicting Polyadenylation Sites from DNA Sequences, Bulk RNA-seq, and Single-cell RNA-seq","authors":"Wenbin Ye , Qiwei Lian , Congting Ye , Xiaohui Wu","doi":"10.1016/j.gpb.2022.09.005","DOIUrl":null,"url":null,"abstract":"<div><p>Alternative <strong>polyadenylation</strong> (APA) plays important roles in modulating mRNA stability, translation, and subcellular localization, and contributes extensively to shaping eukaryotic transcriptome complexity and proteome diversity. Identification of poly(A) sites (pAs) on a genome-wide scale is a critical step toward understanding the underlying mechanism of APA-mediated gene regulation. A number of established computational tools have been proposed to predict pAs from diverse genomic data. Here we provided an exhaustive overview of computational approaches for predicting pAs from DNA sequences, bulk RNA sequencing (<strong>RNA-seq</strong>) data, and single-cell RNA sequencing (<strong>scRNA-seq</strong>) data. Particularly, we examined several representative tools using bulk RNA-seq and scRNA-seq data from peripheral blood mononuclear cells and put forward operable suggestions on how to assess the reliability of pAs predicted by different tools. We also proposed practical guidelines on choosing appropriate methods applicable to diverse scenarios. Moreover, we discussed in depth the challenges in improving the performance of pA prediction and benchmarking different methods. Additionally, we highlighted outstanding challenges and opportunities using new <strong>machine learning</strong> and integrative multi-omics techniques, and provided our perspective on how computational methodologies might evolve in the future for non-3′ untranslated region, tissue-specific, cross-species, and single-cell pA prediction.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"21 1","pages":"Pages 67-83"},"PeriodicalIF":7.9000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ff/97/main.PMC10372920.pdf","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1672022922001218","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/24 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 6
Abstract
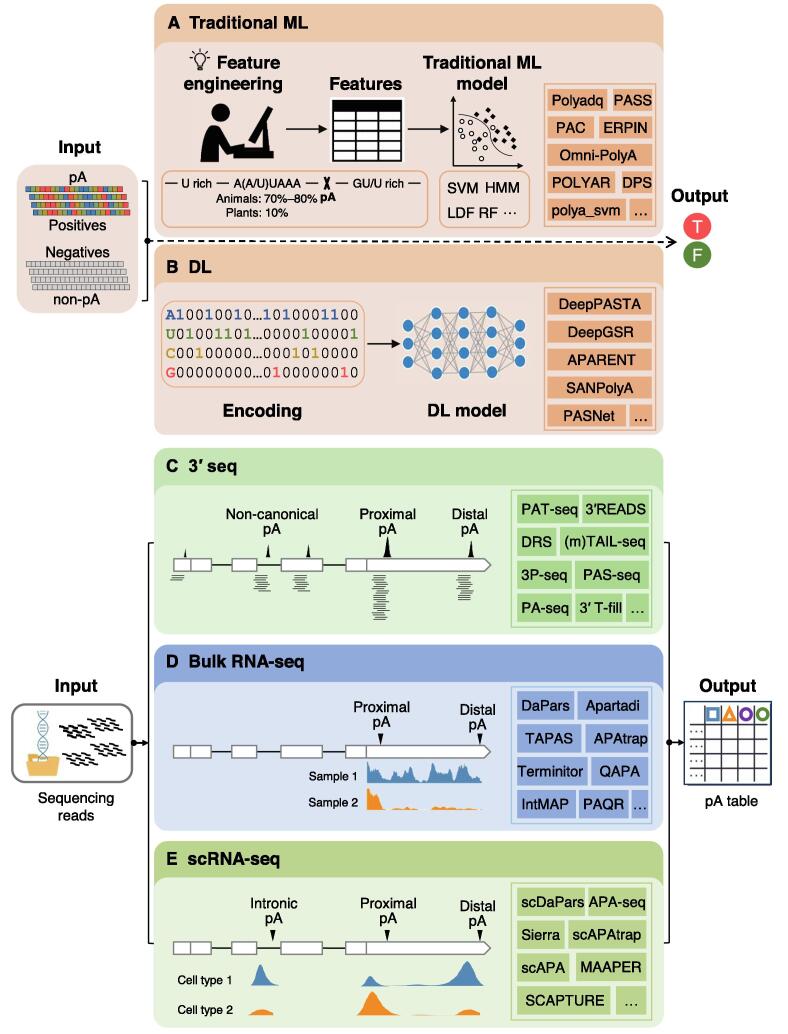
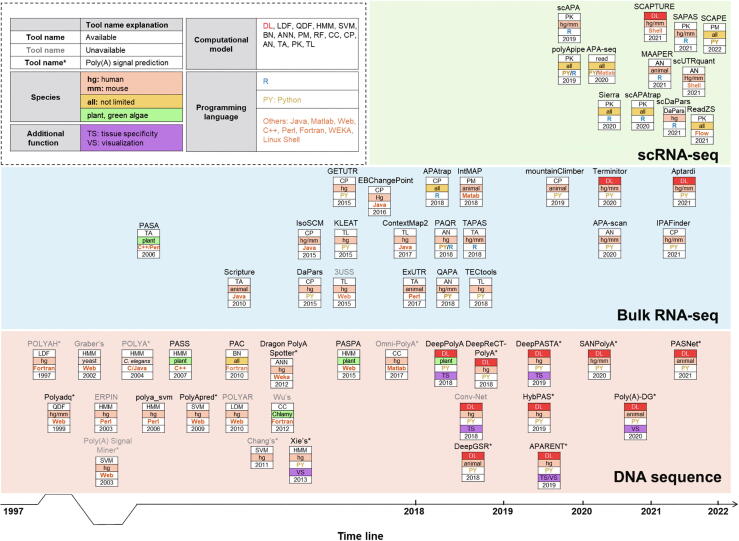
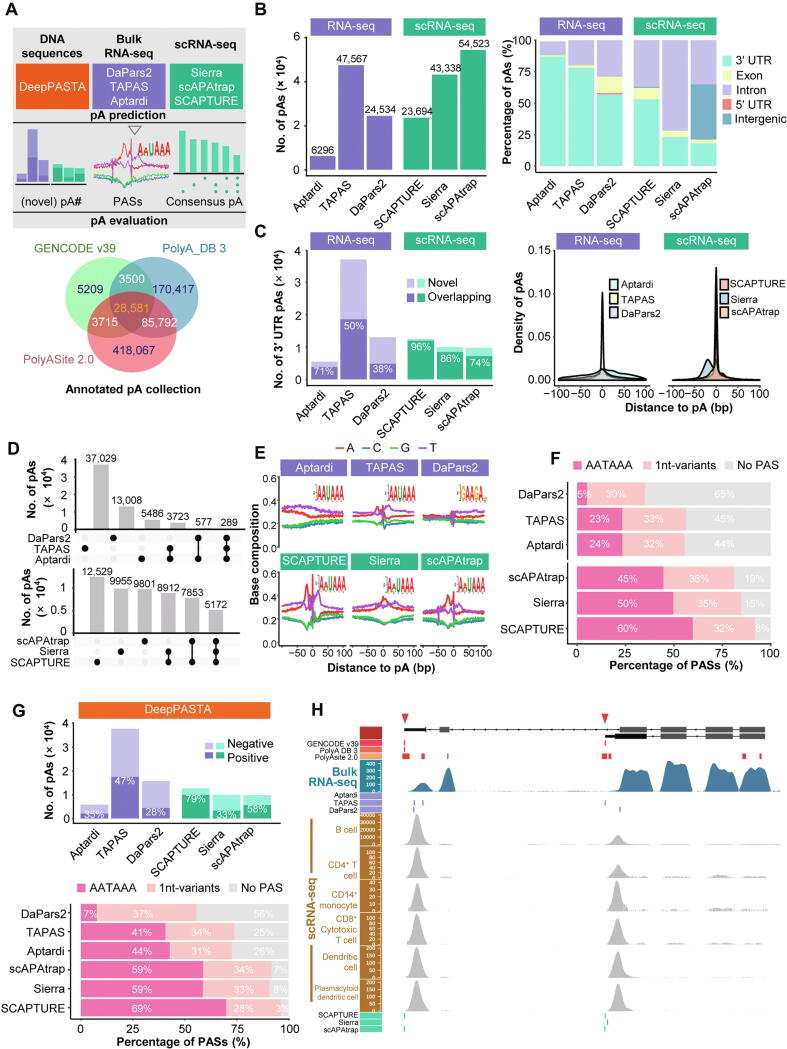
Alternative polyadenylation (APA) plays important roles in modulating mRNA stability, translation, and subcellular localization, and contributes extensively to shaping eukaryotic transcriptome complexity and proteome diversity. Identification of poly(A) sites (pAs) on a genome-wide scale is a critical step toward understanding the underlying mechanism of APA-mediated gene regulation. A number of established computational tools have been proposed to predict pAs from diverse genomic data. Here we provided an exhaustive overview of computational approaches for predicting pAs from DNA sequences, bulk RNA sequencing (RNA-seq) data, and single-cell RNA sequencing (scRNA-seq) data. Particularly, we examined several representative tools using bulk RNA-seq and scRNA-seq data from peripheral blood mononuclear cells and put forward operable suggestions on how to assess the reliability of pAs predicted by different tools. We also proposed practical guidelines on choosing appropriate methods applicable to diverse scenarios. Moreover, we discussed in depth the challenges in improving the performance of pA prediction and benchmarking different methods. Additionally, we highlighted outstanding challenges and opportunities using new machine learning and integrative multi-omics techniques, and provided our perspective on how computational methodologies might evolve in the future for non-3′ untranslated region, tissue-specific, cross-species, and single-cell pA prediction.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
