Robert M Geraghty, Sarah Orr, Eric Olinger, Ruxandra Neatu, Miguel Barroso-Gil, Holly Mabillard, Genomics England Research Consortium, Ian Wilson, John A Sayer
{"title":"Use of whole genome sequencing to determine the genetic basis of visceral myopathies including Prune Belly syndrome.","authors":"Robert M Geraghty, Sarah Orr, Eric Olinger, Ruxandra Neatu, Miguel Barroso-Gil, Holly Mabillard, Genomics England Research Consortium, Ian Wilson, John A Sayer","doi":"10.1007/s44162-023-00012-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives/aims: </strong>The visceral myopathies (VM) are a group of disorders characterised by poorly contractile or acontractile smooth muscle. They manifest in both the GI and GU tracts, ranging from megacystis to Prune Belly syndrome. We aimed to apply a bespoke virtual genetic panel and describe novel variants associated with this condition using whole genome sequencing data within the Genomics England 100,000 Genomes Project.</p><p><strong>Methods: </strong>We screened the Genomics England 100,000 Genomes Project rare diseases database for patients with VM-related phenotypes. These patients were screened for sequence variants and copy number variants (CNV) in <i>ACTG2</i>, <i>ACTA2</i>, <i>MYH11</i>, <i>MYLK</i>, <i>LMOD1</i>, <i>CHRM3</i>, <i>MYL9</i>, <i>FLNA</i> and <i>KNCMA1</i> by analysing whole genome sequencing data. The identified variants were analysed using variant effect predictor online tool, and any possible segregation in other family members and novel missense mutations was modelled using in silico tools. The VM cohort was also used to perform a genome-wide variant burden test in order to identify confirm gene associations in this cohort.</p><p><strong>Results: </strong>We identified 76 patients with phenotypes consistent with a diagnosis of VM. The range of presentations included megacystis/microcolon hypoperistalsis syndrome, Prune Belly syndrome and chronic intestinal pseudo-obstruction. Of the patients in whom we identified heterozygous <i>ACTG2</i> variants, 7 had likely pathogenic variants including 1 novel likely pathogenic allele. There were 4 patients in whom we identified a heterozygous <i>MYH11</i> variant of uncertain significance which leads to a frameshift and a predicted protein elongation. We identified one family in whom we found a heterozygous variant of uncertain significance in <i>KCNMA1</i> which in silico models predicted to be disease causing and may explain the VM phenotype seen. We did not find any CNV changes in known genes leading to VM-related disease phenotypes. In this phenotype selected cohort, <i>ACTG2</i> is the largest monogenic cause of VM-related disease accounting for 9% of the cohort, supported by a variant burden test approach, which identified <i>ACTG2</i> variants as the largest contributor to VM-related phenotypes.</p><p><strong>Conclusions: </strong>VM are a group of disorders that are not easily classified and may be given different diagnostic labels depending on their phenotype. Molecular genetic analysis of these patients is valuable as it allows precise diagnosis and aids understanding of the underlying disease manifestations. We identified <i>ACTG2</i> as the most frequent genetic cause of VM. We recommend a nomenclature change to 'autosomal dominant ACTG2 visceral myopathy' for patients with pathogenic variants in <i>ACTG2</i> and associated VM phenotype<i>s</i>.</p><p><strong>Supplementary information: </strong>The online version contains supplementary material available at 10.1007/s44162-023-00012-z.</p>","PeriodicalId":73925,"journal":{"name":"Journal of rare diseases (Berlin, Germany)","volume":"2 1","pages":"9"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10241726/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of rare diseases (Berlin, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s44162-023-00012-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/5 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives/aims: The visceral myopathies (VM) are a group of disorders characterised by poorly contractile or acontractile smooth muscle. They manifest in both the GI and GU tracts, ranging from megacystis to Prune Belly syndrome. We aimed to apply a bespoke virtual genetic panel and describe novel variants associated with this condition using whole genome sequencing data within the Genomics England 100,000 Genomes Project.
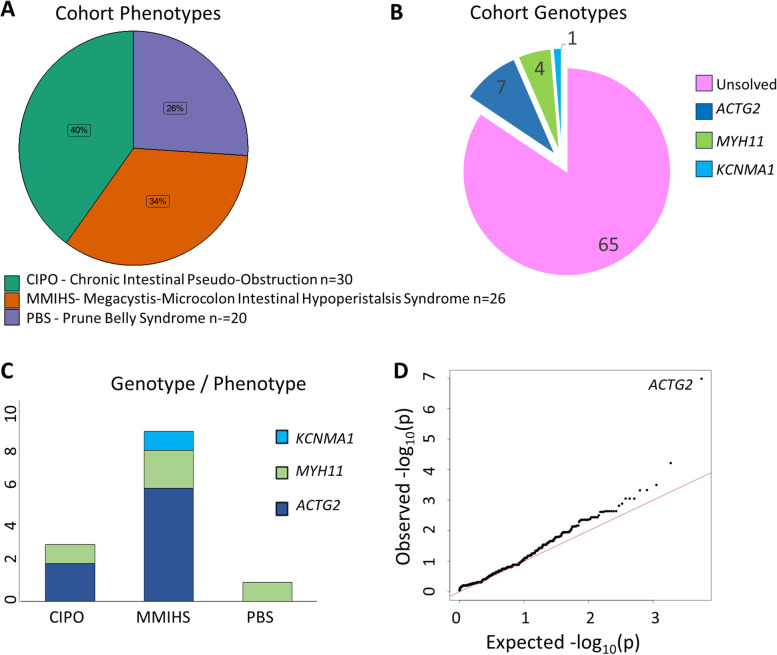
Methods: We screened the Genomics England 100,000 Genomes Project rare diseases database for patients with VM-related phenotypes. These patients were screened for sequence variants and copy number variants (CNV) in ACTG2, ACTA2, MYH11, MYLK, LMOD1, CHRM3, MYL9, FLNA and KNCMA1 by analysing whole genome sequencing data. The identified variants were analysed using variant effect predictor online tool, and any possible segregation in other family members and novel missense mutations was modelled using in silico tools. The VM cohort was also used to perform a genome-wide variant burden test in order to identify confirm gene associations in this cohort.
Results: We identified 76 patients with phenotypes consistent with a diagnosis of VM. The range of presentations included megacystis/microcolon hypoperistalsis syndrome, Prune Belly syndrome and chronic intestinal pseudo-obstruction. Of the patients in whom we identified heterozygous ACTG2 variants, 7 had likely pathogenic variants including 1 novel likely pathogenic allele. There were 4 patients in whom we identified a heterozygous MYH11 variant of uncertain significance which leads to a frameshift and a predicted protein elongation. We identified one family in whom we found a heterozygous variant of uncertain significance in KCNMA1 which in silico models predicted to be disease causing and may explain the VM phenotype seen. We did not find any CNV changes in known genes leading to VM-related disease phenotypes. In this phenotype selected cohort, ACTG2 is the largest monogenic cause of VM-related disease accounting for 9% of the cohort, supported by a variant burden test approach, which identified ACTG2 variants as the largest contributor to VM-related phenotypes.
Conclusions: VM are a group of disorders that are not easily classified and may be given different diagnostic labels depending on their phenotype. Molecular genetic analysis of these patients is valuable as it allows precise diagnosis and aids understanding of the underlying disease manifestations. We identified ACTG2 as the most frequent genetic cause of VM. We recommend a nomenclature change to 'autosomal dominant ACTG2 visceral myopathy' for patients with pathogenic variants in ACTG2 and associated VM phenotypes.
Supplementary information: The online version contains supplementary material available at 10.1007/s44162-023-00012-z.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
