Gülçin Yegen, Ali Yılmaz Altay, İsmail Yılmaz, Yalın İşcan, İsmail Cem Sormaz, Nihat Aksakal, Semen Önder, Özgür Mete
{"title":"DICER1 Mutations Do Not Always Indicate Dismal Prognosis in Pediatric Poorly Differentiated Thyroid Carcinomas.","authors":"Gülçin Yegen, Ali Yılmaz Altay, İsmail Yılmaz, Yalın İşcan, İsmail Cem Sormaz, Nihat Aksakal, Semen Önder, Özgür Mete","doi":"10.1007/s12022-023-09780-2","DOIUrl":null,"url":null,"abstract":"<p><p>Progress in the field of pediatric thyroid pathology has linked DICER1 mutations to benign follicular cell-derived thyroid tumors (e.g., follicular adenoma with papillary architecture, follicular nodular disease), low-risk follicular cell-derived differentiated thyroid carcinomas and PDTCs enriched in fatal or recurrent/progressive disease. The dismal outcome of DICER1-harboring pediatric PDTCs stems from a limited number of reported patients' data given the rarity of pediatric PDTCs. In light of the former observations, the current study assessed clinicopathological variables of a series of 5 pediatric (≤ 18 years old) PDTCs using the Turin criteria (WHO 2022) and also examined the status of DICER1 and TERT promoter mutations. Five PDTCs (3 males, 2 females) were included in the study. The mean age at the time of diagnosis was 15.4 years. No patients had a history of DICER1 syndrome-related tumors or other clinicopathological diagnostic features of DICER1 syndrome. The mean tumor size was 3.9 cm. All tumors were completely submitted for microscopic examination. There was increased mitotic activity ranging from 3 to 10 mitoses per 2 mm<sup>2</sup>. Tumor necrosis was present in two cases. No PDTC harbored TERT promoter mutation. DICER1 hot spot mutation was identified in one (20%) tumor. The DICER1-mutant tumor had neither associated differentiated thyroid carcinoma component nor other pathological findings in the adjacent thyroid parenchyma. The DICER1-mutant PDTC showed widely invasive growth confined to the thyroid parenchyma. Despite the widely invasive growth, the tumor lacked vascular invasion. Two DICER1 wild-type PDTCs had lymphocytic thyroiditis and another one had underlying follicular nodular disease and/or follicular adenomas. Three DICER1 wild-type PDTCs also had an associated differentiated thyroid carcinoma component with no high-grade features. No abnormal p53 expression (overexpression or global loss) was recorded in all tested tumors. Four patients had follow-up data with a mean follow-up time of 60.25 months (range: 18-86 months). One patient with no evidence of disease recurrence died of an unrelated cause after 18 months of the initial surgery, all remaining patients were alive with no distant metastasis at their last visit. Of the 4 patients with lymph node (LN) dissection, one DICER1 wild-type PDTC had recurrent nodal disease. During the follow-up period (72 months), no local recurrence or distant metastases was detected in the DICER1-mutant PDTC. Taken together all reported findings from earlier series, DICER1 mutations alone may not necessarily indicate dismal outcome in a subset of pediatric PDTCs. The occurrence of additional genomic alterations as discussed in some earlier reports may be contributing to tumor progression or aggressivity of pediatric PDTCs. The lack of vascular invasion in the current DICER1-mutant pediatric PDTC may also explain an indolent biologic outcome. The risk escalation of DICER1 mutations should integrate the status of additional genetic events and well-established pathologic variables in order to ensure predictive dynamic risk stratification in DICER1-mutant pediatric PDTCs. Additional studies are needed to corroborate the findings of this study and advance our knowledge in pediatric thyroid neoplasia.</p>","PeriodicalId":55167,"journal":{"name":"Endocrine Pathology","volume":" ","pages":"279-286"},"PeriodicalIF":14.7000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine Pathology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12022-023-09780-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/14 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
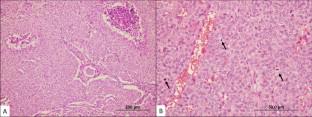
Progress in the field of pediatric thyroid pathology has linked DICER1 mutations to benign follicular cell-derived thyroid tumors (e.g., follicular adenoma with papillary architecture, follicular nodular disease), low-risk follicular cell-derived differentiated thyroid carcinomas and PDTCs enriched in fatal or recurrent/progressive disease. The dismal outcome of DICER1-harboring pediatric PDTCs stems from a limited number of reported patients' data given the rarity of pediatric PDTCs. In light of the former observations, the current study assessed clinicopathological variables of a series of 5 pediatric (≤ 18 years old) PDTCs using the Turin criteria (WHO 2022) and also examined the status of DICER1 and TERT promoter mutations. Five PDTCs (3 males, 2 females) were included in the study. The mean age at the time of diagnosis was 15.4 years. No patients had a history of DICER1 syndrome-related tumors or other clinicopathological diagnostic features of DICER1 syndrome. The mean tumor size was 3.9 cm. All tumors were completely submitted for microscopic examination. There was increased mitotic activity ranging from 3 to 10 mitoses per 2 mm2. Tumor necrosis was present in two cases. No PDTC harbored TERT promoter mutation. DICER1 hot spot mutation was identified in one (20%) tumor. The DICER1-mutant tumor had neither associated differentiated thyroid carcinoma component nor other pathological findings in the adjacent thyroid parenchyma. The DICER1-mutant PDTC showed widely invasive growth confined to the thyroid parenchyma. Despite the widely invasive growth, the tumor lacked vascular invasion. Two DICER1 wild-type PDTCs had lymphocytic thyroiditis and another one had underlying follicular nodular disease and/or follicular adenomas. Three DICER1 wild-type PDTCs also had an associated differentiated thyroid carcinoma component with no high-grade features. No abnormal p53 expression (overexpression or global loss) was recorded in all tested tumors. Four patients had follow-up data with a mean follow-up time of 60.25 months (range: 18-86 months). One patient with no evidence of disease recurrence died of an unrelated cause after 18 months of the initial surgery, all remaining patients were alive with no distant metastasis at their last visit. Of the 4 patients with lymph node (LN) dissection, one DICER1 wild-type PDTC had recurrent nodal disease. During the follow-up period (72 months), no local recurrence or distant metastases was detected in the DICER1-mutant PDTC. Taken together all reported findings from earlier series, DICER1 mutations alone may not necessarily indicate dismal outcome in a subset of pediatric PDTCs. The occurrence of additional genomic alterations as discussed in some earlier reports may be contributing to tumor progression or aggressivity of pediatric PDTCs. The lack of vascular invasion in the current DICER1-mutant pediatric PDTC may also explain an indolent biologic outcome. The risk escalation of DICER1 mutations should integrate the status of additional genetic events and well-established pathologic variables in order to ensure predictive dynamic risk stratification in DICER1-mutant pediatric PDTCs. Additional studies are needed to corroborate the findings of this study and advance our knowledge in pediatric thyroid neoplasia.
期刊介绍:
Endocrine Pathology publishes original articles on clinical and basic aspects of endocrine disorders. Work with animals or in vitro techniques is acceptable if it is relevant to human normal or abnormal endocrinology. Manuscripts will be considered for publication in the form of original articles, case reports, clinical case presentations, reviews, and descriptions of techniques. Submission of a paper implies that it reports unpublished work, except in abstract form, and is not being submitted simultaneously to another publication. Accepted manuscripts become the sole property of Endocrine Pathology and may not be published elsewhere without written consent from the publisher. All articles are subject to review by experienced referees. The Editors and Editorial Board judge manuscripts suitable for publication, and decisions by the Editors are final.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
