{"title":"引起视网膜色素变性或Usher综合征的USH2A变体在疾病特异性类器官中引起不同的视网膜表型。","authors":"Carla Sanjurjo-Soriano, Carla Jimenez-Medina, Nejla Erkilic, Luisina Cappellino, Arnaud Lefevre, Kerstin Nagel-Wolfrum, Uwe Wolfrum, Erwin Van Wijk, Anne-Françoise Roux, Isabelle Meunier, Vasiliki Kalatzis","doi":"10.1016/j.xhgg.2023.100229","DOIUrl":null,"url":null,"abstract":"<p><p>There is an emblematic clinical and genetic heterogeneity associated with inherited retinal diseases (IRDs). The most common form is retinitis pigmentosa (RP), a rod-cone dystrophy caused by pathogenic variants in over 80 different genes. Further complexifying diagnosis, different variants in individual RP genes can also alter the clinical phenotype. <i>USH2A</i> is the most prevalent gene for autosomal-recessive RP and one of the most challenging because of its large size and, hence, large number of variants. Moreover, <i>USH2A</i> variants give rise to non-syndromic and syndromic RP, known as Usher syndrome (USH) type 2, which is associated with vision and hearing loss. The lack of a clear genotype-phenotype correlation or prognostic models renders diagnosis highly challenging. We report here a long-awaited differential non-syndromic RP and USH phenotype in three human disease-specific models: fibroblasts, induced pluripotent stem cells (iPSCs), and mature iPSC-derived retinal organoids. Moreover, we identified distinct retinal phenotypes in organoids from multiple RP and USH individuals, which were validated by isogenic-corrected controls. Non-syndromic RP organoids showed compromised photoreceptor differentiation, whereas USH organoids showed a striking and unexpected cone phenotype. Furthermore, complementary clinical investigations identified macular atrophy in a high proportion of USH compared with RP individuals, further validating our observations that <i>USH2A</i> variants differentially affect cones. Overall, identification of distinct non-syndromic RP and USH phenotypes in multiple models provides valuable and robust readouts for testing the pathogenicity of <i>USH2A</i> variants as well as the efficacy of therapeutic approaches in complementary cell types.</p>","PeriodicalId":34530,"journal":{"name":"HGG Advances","volume":"4 4","pages":"100229"},"PeriodicalIF":3.6000,"publicationDate":"2023-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/15/42/main.PMC10465966.pdf","citationCount":"0","resultStr":"{\"title\":\"<i>USH2A</i> variants causing retinitis pigmentosa or Usher syndrome provoke differential retinal phenotypes in disease-specific organoids.\",\"authors\":\"Carla Sanjurjo-Soriano, Carla Jimenez-Medina, Nejla Erkilic, Luisina Cappellino, Arnaud Lefevre, Kerstin Nagel-Wolfrum, Uwe Wolfrum, Erwin Van Wijk, Anne-Françoise Roux, Isabelle Meunier, Vasiliki Kalatzis\",\"doi\":\"10.1016/j.xhgg.2023.100229\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>There is an emblematic clinical and genetic heterogeneity associated with inherited retinal diseases (IRDs). The most common form is retinitis pigmentosa (RP), a rod-cone dystrophy caused by pathogenic variants in over 80 different genes. Further complexifying diagnosis, different variants in individual RP genes can also alter the clinical phenotype. <i>USH2A</i> is the most prevalent gene for autosomal-recessive RP and one of the most challenging because of its large size and, hence, large number of variants. Moreover, <i>USH2A</i> variants give rise to non-syndromic and syndromic RP, known as Usher syndrome (USH) type 2, which is associated with vision and hearing loss. The lack of a clear genotype-phenotype correlation or prognostic models renders diagnosis highly challenging. We report here a long-awaited differential non-syndromic RP and USH phenotype in three human disease-specific models: fibroblasts, induced pluripotent stem cells (iPSCs), and mature iPSC-derived retinal organoids. Moreover, we identified distinct retinal phenotypes in organoids from multiple RP and USH individuals, which were validated by isogenic-corrected controls. Non-syndromic RP organoids showed compromised photoreceptor differentiation, whereas USH organoids showed a striking and unexpected cone phenotype. Furthermore, complementary clinical investigations identified macular atrophy in a high proportion of USH compared with RP individuals, further validating our observations that <i>USH2A</i> variants differentially affect cones. Overall, identification of distinct non-syndromic RP and USH phenotypes in multiple models provides valuable and robust readouts for testing the pathogenicity of <i>USH2A</i> variants as well as the efficacy of therapeutic approaches in complementary cell types.</p>\",\"PeriodicalId\":34530,\"journal\":{\"name\":\"HGG Advances\",\"volume\":\"4 4\",\"pages\":\"100229\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2023-08-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/15/42/main.PMC10465966.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"HGG Advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1016/j.xhgg.2023.100229\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/12 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"HGG Advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.xhgg.2023.100229","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/12 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
USH2A variants causing retinitis pigmentosa or Usher syndrome provoke differential retinal phenotypes in disease-specific organoids.
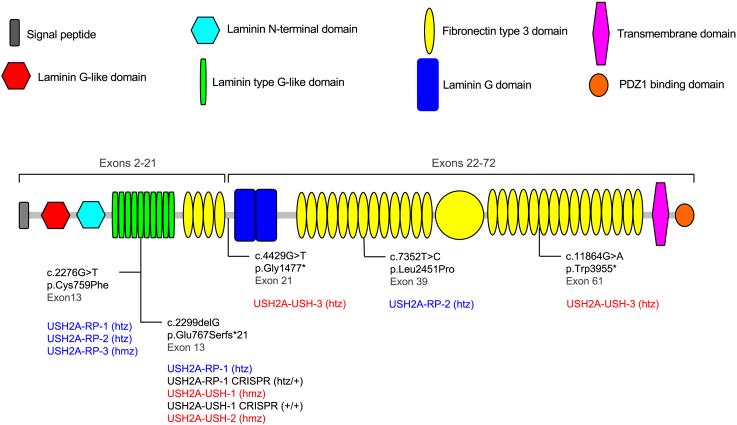
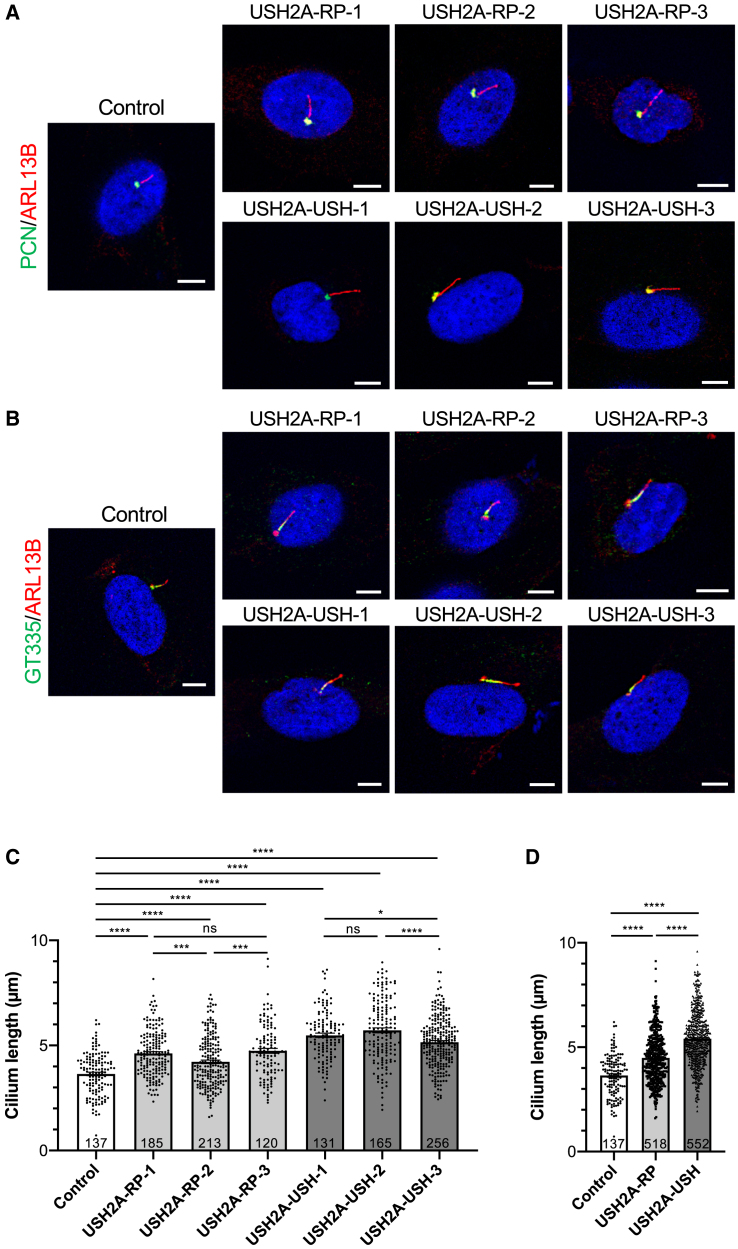
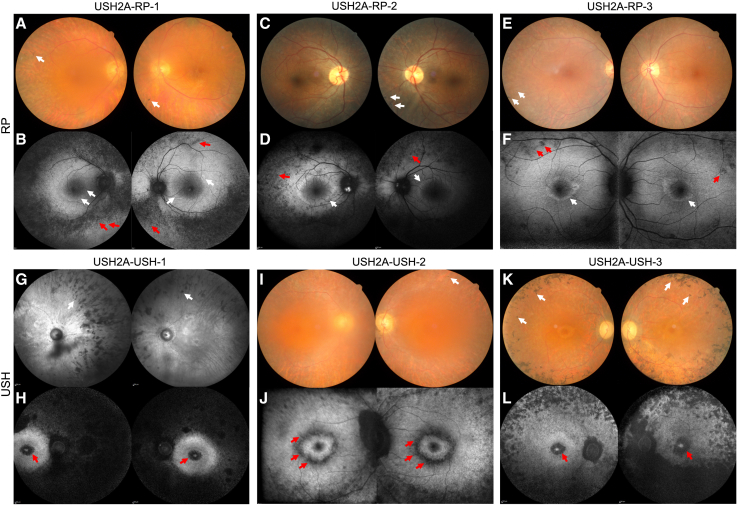
There is an emblematic clinical and genetic heterogeneity associated with inherited retinal diseases (IRDs). The most common form is retinitis pigmentosa (RP), a rod-cone dystrophy caused by pathogenic variants in over 80 different genes. Further complexifying diagnosis, different variants in individual RP genes can also alter the clinical phenotype. USH2A is the most prevalent gene for autosomal-recessive RP and one of the most challenging because of its large size and, hence, large number of variants. Moreover, USH2A variants give rise to non-syndromic and syndromic RP, known as Usher syndrome (USH) type 2, which is associated with vision and hearing loss. The lack of a clear genotype-phenotype correlation or prognostic models renders diagnosis highly challenging. We report here a long-awaited differential non-syndromic RP and USH phenotype in three human disease-specific models: fibroblasts, induced pluripotent stem cells (iPSCs), and mature iPSC-derived retinal organoids. Moreover, we identified distinct retinal phenotypes in organoids from multiple RP and USH individuals, which were validated by isogenic-corrected controls. Non-syndromic RP organoids showed compromised photoreceptor differentiation, whereas USH organoids showed a striking and unexpected cone phenotype. Furthermore, complementary clinical investigations identified macular atrophy in a high proportion of USH compared with RP individuals, further validating our observations that USH2A variants differentially affect cones. Overall, identification of distinct non-syndromic RP and USH phenotypes in multiple models provides valuable and robust readouts for testing the pathogenicity of USH2A variants as well as the efficacy of therapeutic approaches in complementary cell types.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
