{"title":"在atm缺陷肿瘤细胞中,依托泊苷在靶向抑制DNA-PKcs过程中诱导的选择性末端连接产生稳定的染色体畸变。","authors":"Marcelo de Campos Nebel, Micaela Palmitelli, Josefina Pérez Maturo, Marcela González-Cid","doi":"10.1007/s10577-022-09700-w","DOIUrl":null,"url":null,"abstract":"<p><p>ATM and DNA-PKcs coordinate the DNA damage response at multiple levels following the exposure to chemotherapy. The Topoisomerase II poison etoposide (ETO) is an effective chemotherapeutic agent that induces DNA double-strand breaks (DSB), but it is responsible from the chromosomal rearrangements frequently found in therapy-related secondary tumors. Targeted inhibition of DNA-PKcs in ATM-defective tumors combined with radio- or chemotherapy has been proposed as relevant therapies. Here, we explored the DNA repair mechanisms and the genetic consequences of targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumor cells after exposure to ETO. We demonstrated that chemical inhibition of DNA-PKcs followed by treatment with ETO resulted in the accumulation of chromatid breaks and decreased mitotic index in both A-T cells and ATM-knocked-down (ATM<sup>kd</sup>) tumor cells. The HR repair process in DNA-PKcs-inhibited ATM<sup>kd</sup> cells amplified the RAD51 foci number, with no correlated increase in sister chromatid exchanges. The analysis of post-mitotic DNA lesions presented an augmented number of persistent unresolved DSB, without alterations in the cell cycle progression. Long-term examination of chromosome aberrations revealed a strikingly high number of chromatid and chromosome exchanges. By using genetic and pharmacological abrogation of PARP-1, we demonstrated that alternative end-joining (alt-EJ) repair pathway is responsible for those chromosome abnormalities generated by limiting c-NHEJ activities during directed inhibition of DNA-PKcs in ATM-deficient cells. Targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumors stimulates the DSB repair by alt-EJ, which is liable for the origin of cells carrying stable chromosome aberrations that may eventually restrict the therapeutic strategy.</p>","PeriodicalId":50698,"journal":{"name":"Chromosome Research","volume":"30 4","pages":"459-476"},"PeriodicalIF":2.8000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Alternative end-joining originates stable chromosome aberrations induced by etoposide during targeted inhibition of DNA-PKcs in ATM-deficient tumor cells.\",\"authors\":\"Marcelo de Campos Nebel, Micaela Palmitelli, Josefina Pérez Maturo, Marcela González-Cid\",\"doi\":\"10.1007/s10577-022-09700-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>ATM and DNA-PKcs coordinate the DNA damage response at multiple levels following the exposure to chemotherapy. The Topoisomerase II poison etoposide (ETO) is an effective chemotherapeutic agent that induces DNA double-strand breaks (DSB), but it is responsible from the chromosomal rearrangements frequently found in therapy-related secondary tumors. Targeted inhibition of DNA-PKcs in ATM-defective tumors combined with radio- or chemotherapy has been proposed as relevant therapies. Here, we explored the DNA repair mechanisms and the genetic consequences of targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumor cells after exposure to ETO. We demonstrated that chemical inhibition of DNA-PKcs followed by treatment with ETO resulted in the accumulation of chromatid breaks and decreased mitotic index in both A-T cells and ATM-knocked-down (ATM<sup>kd</sup>) tumor cells. The HR repair process in DNA-PKcs-inhibited ATM<sup>kd</sup> cells amplified the RAD51 foci number, with no correlated increase in sister chromatid exchanges. The analysis of post-mitotic DNA lesions presented an augmented number of persistent unresolved DSB, without alterations in the cell cycle progression. Long-term examination of chromosome aberrations revealed a strikingly high number of chromatid and chromosome exchanges. By using genetic and pharmacological abrogation of PARP-1, we demonstrated that alternative end-joining (alt-EJ) repair pathway is responsible for those chromosome abnormalities generated by limiting c-NHEJ activities during directed inhibition of DNA-PKcs in ATM-deficient cells. Targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumors stimulates the DSB repair by alt-EJ, which is liable for the origin of cells carrying stable chromosome aberrations that may eventually restrict the therapeutic strategy.</p>\",\"PeriodicalId\":50698,\"journal\":{\"name\":\"Chromosome Research\",\"volume\":\"30 4\",\"pages\":\"459-476\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chromosome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s10577-022-09700-w\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chromosome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s10577-022-09700-w","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Alternative end-joining originates stable chromosome aberrations induced by etoposide during targeted inhibition of DNA-PKcs in ATM-deficient tumor cells.
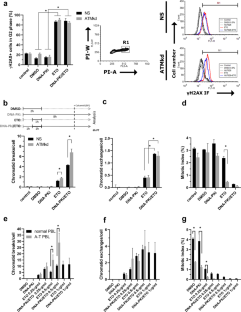
ATM and DNA-PKcs coordinate the DNA damage response at multiple levels following the exposure to chemotherapy. The Topoisomerase II poison etoposide (ETO) is an effective chemotherapeutic agent that induces DNA double-strand breaks (DSB), but it is responsible from the chromosomal rearrangements frequently found in therapy-related secondary tumors. Targeted inhibition of DNA-PKcs in ATM-defective tumors combined with radio- or chemotherapy has been proposed as relevant therapies. Here, we explored the DNA repair mechanisms and the genetic consequences of targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumor cells after exposure to ETO. We demonstrated that chemical inhibition of DNA-PKcs followed by treatment with ETO resulted in the accumulation of chromatid breaks and decreased mitotic index in both A-T cells and ATM-knocked-down (ATMkd) tumor cells. The HR repair process in DNA-PKcs-inhibited ATMkd cells amplified the RAD51 foci number, with no correlated increase in sister chromatid exchanges. The analysis of post-mitotic DNA lesions presented an augmented number of persistent unresolved DSB, without alterations in the cell cycle progression. Long-term examination of chromosome aberrations revealed a strikingly high number of chromatid and chromosome exchanges. By using genetic and pharmacological abrogation of PARP-1, we demonstrated that alternative end-joining (alt-EJ) repair pathway is responsible for those chromosome abnormalities generated by limiting c-NHEJ activities during directed inhibition of DNA-PKcs in ATM-deficient cells. Targeting the non-oncogenic addiction to DNA-PKcs of ATM-defective tumors stimulates the DSB repair by alt-EJ, which is liable for the origin of cells carrying stable chromosome aberrations that may eventually restrict the therapeutic strategy.
期刊介绍:
Chromosome Research publishes manuscripts from work based on all organisms and encourages submissions in the following areas including, but not limited, to:
· Chromosomes and their linkage to diseases;
· Chromosome organization within the nucleus;
· Chromatin biology (transcription, non-coding RNA, etc);
· Chromosome structure, function and mechanics;
· Chromosome and DNA repair;
· Epigenetic chromosomal functions (centromeres, telomeres, replication, imprinting,
dosage compensation, sex determination, chromosome remodeling);
· Architectural/epigenomic organization of the genome;
· Functional annotation of the genome;
· Functional and comparative genomics in plants and animals;
· Karyology studies that help resolve difficult taxonomic problems or that provide
clues to fundamental mechanisms of genome and karyotype evolution in plants and animals;
· Mitosis and Meiosis;
· Cancer cytogenomics.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
