Serena Aneli, Piero Fariselli, Elena Chierto, Carla Bini, Carlo Robino, Giovanni Birolo
{"title":"Recombulator-X:一种快速且用户友好的工具,用于估计法医遗传学中X染色体重组率。","authors":"Serena Aneli, Piero Fariselli, Elena Chierto, Carla Bini, Carlo Robino, Giovanni Birolo","doi":"10.1371/journal.pcbi.1011474","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic markers (especially short tandem repeats or STRs) located on the X chromosome are a valuable resource to solve complex kinship cases in forensic genetics in addition or alternatively to autosomal STRs. Groups of tightly linked markers are combined into haplotypes, thus increasing the discriminating power of tests. However, this approach requires precise knowledge of the recombination rates between adjacent markers. The International Society of Forensic Genetics recommends that recombination rate estimation on the X chromosome is performed from pedigree genetic data while taking into account the confounding effect of mutations. However, implementations that satisfy these requirements have several drawbacks: they were never publicly released, they are very slow and/or need cluster-level hardware and strong computational expertise to use. In order to address these key concerns we developed Recombulator-X, a new open-source Python tool. The most challenging issue, namely the running time, was addressed with dynamic programming techniques to greatly reduce the computational complexity of the algorithm. Compared to the previous methods, Recombulator-X reduces the estimation times from weeks or months to less than one hour for typical datasets. Moreover, the estimation process, including preprocessing, has been streamlined and packaged into a simple command-line tool that can be run on a normal PC. Where previous approaches were limited to small panels of STR markers (up to 15), our tool can handle greater numbers (up to 100) of mixed STR and non-STR markers. In conclusion, Recombulator-X makes the estimation process much simpler, faster and accessible to researchers without a computational background, hopefully spurring increased adoption of best practices.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":"19 9","pages":"e1011474"},"PeriodicalIF":4.3000,"publicationDate":"2023-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10538763/pdf/","citationCount":"0","resultStr":"{\"title\":\"Recombulator-X: A fast and user-friendly tool for estimating X chromosome recombination rates in forensic genetics.\",\"authors\":\"Serena Aneli, Piero Fariselli, Elena Chierto, Carla Bini, Carlo Robino, Giovanni Birolo\",\"doi\":\"10.1371/journal.pcbi.1011474\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Genetic markers (especially short tandem repeats or STRs) located on the X chromosome are a valuable resource to solve complex kinship cases in forensic genetics in addition or alternatively to autosomal STRs. Groups of tightly linked markers are combined into haplotypes, thus increasing the discriminating power of tests. However, this approach requires precise knowledge of the recombination rates between adjacent markers. The International Society of Forensic Genetics recommends that recombination rate estimation on the X chromosome is performed from pedigree genetic data while taking into account the confounding effect of mutations. However, implementations that satisfy these requirements have several drawbacks: they were never publicly released, they are very slow and/or need cluster-level hardware and strong computational expertise to use. In order to address these key concerns we developed Recombulator-X, a new open-source Python tool. The most challenging issue, namely the running time, was addressed with dynamic programming techniques to greatly reduce the computational complexity of the algorithm. Compared to the previous methods, Recombulator-X reduces the estimation times from weeks or months to less than one hour for typical datasets. Moreover, the estimation process, including preprocessing, has been streamlined and packaged into a simple command-line tool that can be run on a normal PC. Where previous approaches were limited to small panels of STR markers (up to 15), our tool can handle greater numbers (up to 100) of mixed STR and non-STR markers. In conclusion, Recombulator-X makes the estimation process much simpler, faster and accessible to researchers without a computational background, hopefully spurring increased adoption of best practices.</p>\",\"PeriodicalId\":49688,\"journal\":{\"name\":\"PLoS Computational Biology\",\"volume\":\"19 9\",\"pages\":\"e1011474\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2023-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10538763/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PLoS Computational Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1371/journal.pcbi.1011474\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1011474","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Recombulator-X: A fast and user-friendly tool for estimating X chromosome recombination rates in forensic genetics.
Genetic markers (especially short tandem repeats or STRs) located on the X chromosome are a valuable resource to solve complex kinship cases in forensic genetics in addition or alternatively to autosomal STRs. Groups of tightly linked markers are combined into haplotypes, thus increasing the discriminating power of tests. However, this approach requires precise knowledge of the recombination rates between adjacent markers. The International Society of Forensic Genetics recommends that recombination rate estimation on the X chromosome is performed from pedigree genetic data while taking into account the confounding effect of mutations. However, implementations that satisfy these requirements have several drawbacks: they were never publicly released, they are very slow and/or need cluster-level hardware and strong computational expertise to use. In order to address these key concerns we developed Recombulator-X, a new open-source Python tool. The most challenging issue, namely the running time, was addressed with dynamic programming techniques to greatly reduce the computational complexity of the algorithm. Compared to the previous methods, Recombulator-X reduces the estimation times from weeks or months to less than one hour for typical datasets. Moreover, the estimation process, including preprocessing, has been streamlined and packaged into a simple command-line tool that can be run on a normal PC. Where previous approaches were limited to small panels of STR markers (up to 15), our tool can handle greater numbers (up to 100) of mixed STR and non-STR markers. In conclusion, Recombulator-X makes the estimation process much simpler, faster and accessible to researchers without a computational background, hopefully spurring increased adoption of best practices.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.
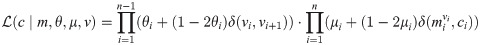
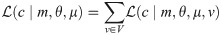



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
