{"title":"红斑狼疮:临床表现、发病机制和治疗方案的最新回顾","authors":"Tejas P. Joshi, Madeleine Duvic","doi":"10.1007/s40257-023-00836-x","DOIUrl":null,"url":null,"abstract":"<div><p>Pityriasis rubra pilaris (PRP) is a rare papulosquamous reaction pattern with a significant impact on quality of life. Type I PRP is the most common PRP variant, presenting as erythematous papules emerging in a follicular distribution and later coalescing into plaques with characteristic islands of sparing; histologically, an alternating pattern of orthokeratosis and parakeratosis is considered the hallmark of PRP (checkerboard hyperkeratosis). Other PRP variants (types II–V) differ in their age of onset and clinical presentation. Type VI PRP is a rare PRP subtype associated with human immunodeficiency virus infection and is occasionally associated with diseases of the follicular occlusion tetrad. <i>Caspase recruitment domain family, member 14</i> (<i>CARD14</i>)-associated papulosquamous eruption and facial discoid dermatitis are newly described disease states that have an important clinical overlap with PRP, creating shared conundrums with respect to diagnosis and treatment. The etiology inciting PRP often remains uncertain; PRP has been suggested to be associated with infection, malignancy, or drug/vaccine administration in some cases, although these are based on case reports and causality has not been established. Type V PRP is often due to inborn <i>CARD14</i> mutations. Furthermore, recent literature has identified interleukin-23/T-helper-17 cell axis dysregulation to be a major mediator of PRP pathogenesis, paving the way for mechanism-directed therapy. At present, high-dose isotretinoin, ixekizumab, and secukinumab are systemic agents supported by single-arm prospective studies; numerous other agents have also been trialed for PRP, with variable success rates. Here, we discuss updates on clinical manifestations, present new insights into etiopathogenesis, and offer a survey of recently described therapeutic options.</p></div>","PeriodicalId":7706,"journal":{"name":"American Journal of Clinical Dermatology","volume":"25 2","pages":"243 - 259"},"PeriodicalIF":8.8000,"publicationDate":"2023-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Pityriasis Rubra Pilaris: An Updated Review of Clinical Presentation, Etiopathogenesis, and Treatment Options\",\"authors\":\"Tejas P. Joshi, Madeleine Duvic\",\"doi\":\"10.1007/s40257-023-00836-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Pityriasis rubra pilaris (PRP) is a rare papulosquamous reaction pattern with a significant impact on quality of life. Type I PRP is the most common PRP variant, presenting as erythematous papules emerging in a follicular distribution and later coalescing into plaques with characteristic islands of sparing; histologically, an alternating pattern of orthokeratosis and parakeratosis is considered the hallmark of PRP (checkerboard hyperkeratosis). Other PRP variants (types II–V) differ in their age of onset and clinical presentation. Type VI PRP is a rare PRP subtype associated with human immunodeficiency virus infection and is occasionally associated with diseases of the follicular occlusion tetrad. <i>Caspase recruitment domain family, member 14</i> (<i>CARD14</i>)-associated papulosquamous eruption and facial discoid dermatitis are newly described disease states that have an important clinical overlap with PRP, creating shared conundrums with respect to diagnosis and treatment. The etiology inciting PRP often remains uncertain; PRP has been suggested to be associated with infection, malignancy, or drug/vaccine administration in some cases, although these are based on case reports and causality has not been established. Type V PRP is often due to inborn <i>CARD14</i> mutations. Furthermore, recent literature has identified interleukin-23/T-helper-17 cell axis dysregulation to be a major mediator of PRP pathogenesis, paving the way for mechanism-directed therapy. At present, high-dose isotretinoin, ixekizumab, and secukinumab are systemic agents supported by single-arm prospective studies; numerous other agents have also been trialed for PRP, with variable success rates. Here, we discuss updates on clinical manifestations, present new insights into etiopathogenesis, and offer a survey of recently described therapeutic options.</p></div>\",\"PeriodicalId\":7706,\"journal\":{\"name\":\"American Journal of Clinical Dermatology\",\"volume\":\"25 2\",\"pages\":\"243 - 259\"},\"PeriodicalIF\":8.8000,\"publicationDate\":\"2023-12-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"American Journal of Clinical Dermatology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s40257-023-00836-x\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"DERMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Clinical Dermatology","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s40257-023-00836-x","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"DERMATOLOGY","Score":null,"Total":0}
Pityriasis Rubra Pilaris: An Updated Review of Clinical Presentation, Etiopathogenesis, and Treatment Options
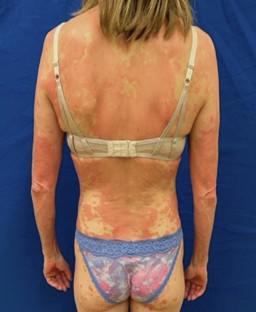
Pityriasis rubra pilaris (PRP) is a rare papulosquamous reaction pattern with a significant impact on quality of life. Type I PRP is the most common PRP variant, presenting as erythematous papules emerging in a follicular distribution and later coalescing into plaques with characteristic islands of sparing; histologically, an alternating pattern of orthokeratosis and parakeratosis is considered the hallmark of PRP (checkerboard hyperkeratosis). Other PRP variants (types II–V) differ in their age of onset and clinical presentation. Type VI PRP is a rare PRP subtype associated with human immunodeficiency virus infection and is occasionally associated with diseases of the follicular occlusion tetrad. Caspase recruitment domain family, member 14 (CARD14)-associated papulosquamous eruption and facial discoid dermatitis are newly described disease states that have an important clinical overlap with PRP, creating shared conundrums with respect to diagnosis and treatment. The etiology inciting PRP often remains uncertain; PRP has been suggested to be associated with infection, malignancy, or drug/vaccine administration in some cases, although these are based on case reports and causality has not been established. Type V PRP is often due to inborn CARD14 mutations. Furthermore, recent literature has identified interleukin-23/T-helper-17 cell axis dysregulation to be a major mediator of PRP pathogenesis, paving the way for mechanism-directed therapy. At present, high-dose isotretinoin, ixekizumab, and secukinumab are systemic agents supported by single-arm prospective studies; numerous other agents have also been trialed for PRP, with variable success rates. Here, we discuss updates on clinical manifestations, present new insights into etiopathogenesis, and offer a survey of recently described therapeutic options.
期刊介绍:
The American Journal of Clinical Dermatology is dedicated to evidence-based therapy and effective patient management in dermatology. It publishes critical review articles and clinically focused original research covering comprehensive aspects of dermatological conditions. The journal enhances visibility and educational value through features like Key Points summaries, plain language summaries, and various digital elements, ensuring accessibility and depth for a diverse readership.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
