Laura Hewson, Amanda Choo, Dani L. Webber, Paul J. Trim, Marten F. Snel, Anthony O. Fedele, John J. Hopwood, Kim M. Hemsley, Louise V. O'Keefe
{"title":"黑腹果蝇的 MPS IIIC(Hgsnat 缺乏症)模型突显了神经胶质细胞在疾病表现中的作用。","authors":"Laura Hewson, Amanda Choo, Dani L. Webber, Paul J. Trim, Marten F. Snel, Anthony O. Fedele, John J. Hopwood, Kim M. Hemsley, Louise V. O'Keefe","doi":"10.1002/jimd.12712","DOIUrl":null,"url":null,"abstract":"<p>Sanfilippo syndrome (Mucopolysaccharidosis type III or MPS III) is a recessively inherited neurodegenerative lysosomal storage disorder. Mutations in genes encoding enzymes in the heparan sulphate degradation pathway lead to the accumulation of partially degraded heparan sulphate, resulting ultimately in the development of neurological deficits. Mutations in the gene encoding the membrane protein heparan-α-glucosaminide <i>N</i>-acetyltransferase (<i>HGSNAT</i>; EC2.3.1.78) cause MPS IIIC (OMIM#252930), typified by impaired cognition, sleep–wake cycle changes, hyperactivity and early death, often before adulthood. The precise disease mechanism that causes symptom emergence remains unknown, posing a significant challenge in the development of effective therapeutics. As <i>HGSNAT</i> is conserved in <i>Drosophila melanogaster</i>, we now describe the creation and characterisation of the first <i>Drosophila</i> models of MPS IIIC. Flies with either an endogenous insertion mutation or RNAi-mediated knockdown of <i>hgsnat</i> were confirmed to have a reduced level of HGSNAT transcripts and age-dependent accumulation of heparan sulphate leading to engorgement of the endo/lysosomal compartment. This resulted in abnormalities at the pre-synapse, defective climbing and reduced overall activity. Altered circadian rhythms (shift in peak morning activity) were seen in <i>hgsnat</i> neuronal knockdown lines. Further, when <i>hgsnat</i> was knocked down in specific glial subsets (wrapping, cortical, astrocytes or subperineural glia), impaired climbing or reduced activity was noted, implying that <i>hgsnat</i> function in these specific glial subtypes contributes significantly to this behaviour and targeting treatments to these cell groups may be necessary to ameliorate or prevent symptom onset. These novel models of MPS IIIC provide critical research tools for delineating the key cellular pathways causal in the onset of neurodegeneration in this presently untreatable disorder.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 2","pages":"340-354"},"PeriodicalIF":3.8000,"publicationDate":"2024-01-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12712","citationCount":"0","resultStr":"{\"title\":\"Drosophila melanogaster models of MPS IIIC (Hgsnat-deficiency) highlight the role of glia in disease presentation\",\"authors\":\"Laura Hewson, Amanda Choo, Dani L. Webber, Paul J. Trim, Marten F. Snel, Anthony O. Fedele, John J. Hopwood, Kim M. Hemsley, Louise V. O'Keefe\",\"doi\":\"10.1002/jimd.12712\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Sanfilippo syndrome (Mucopolysaccharidosis type III or MPS III) is a recessively inherited neurodegenerative lysosomal storage disorder. Mutations in genes encoding enzymes in the heparan sulphate degradation pathway lead to the accumulation of partially degraded heparan sulphate, resulting ultimately in the development of neurological deficits. Mutations in the gene encoding the membrane protein heparan-α-glucosaminide <i>N</i>-acetyltransferase (<i>HGSNAT</i>; EC2.3.1.78) cause MPS IIIC (OMIM#252930), typified by impaired cognition, sleep–wake cycle changes, hyperactivity and early death, often before adulthood. The precise disease mechanism that causes symptom emergence remains unknown, posing a significant challenge in the development of effective therapeutics. As <i>HGSNAT</i> is conserved in <i>Drosophila melanogaster</i>, we now describe the creation and characterisation of the first <i>Drosophila</i> models of MPS IIIC. Flies with either an endogenous insertion mutation or RNAi-mediated knockdown of <i>hgsnat</i> were confirmed to have a reduced level of HGSNAT transcripts and age-dependent accumulation of heparan sulphate leading to engorgement of the endo/lysosomal compartment. This resulted in abnormalities at the pre-synapse, defective climbing and reduced overall activity. Altered circadian rhythms (shift in peak morning activity) were seen in <i>hgsnat</i> neuronal knockdown lines. Further, when <i>hgsnat</i> was knocked down in specific glial subsets (wrapping, cortical, astrocytes or subperineural glia), impaired climbing or reduced activity was noted, implying that <i>hgsnat</i> function in these specific glial subtypes contributes significantly to this behaviour and targeting treatments to these cell groups may be necessary to ameliorate or prevent symptom onset. These novel models of MPS IIIC provide critical research tools for delineating the key cellular pathways causal in the onset of neurodegeneration in this presently untreatable disorder.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 2\",\"pages\":\"340-354\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-01-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12712\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12712\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12712","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Drosophila melanogaster models of MPS IIIC (Hgsnat-deficiency) highlight the role of glia in disease presentation
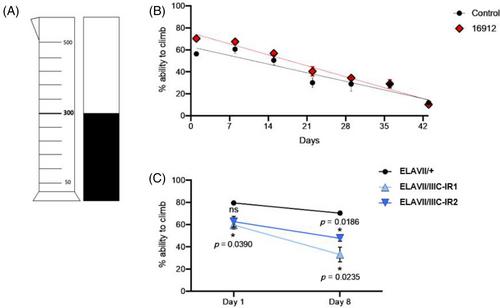
Sanfilippo syndrome (Mucopolysaccharidosis type III or MPS III) is a recessively inherited neurodegenerative lysosomal storage disorder. Mutations in genes encoding enzymes in the heparan sulphate degradation pathway lead to the accumulation of partially degraded heparan sulphate, resulting ultimately in the development of neurological deficits. Mutations in the gene encoding the membrane protein heparan-α-glucosaminide N-acetyltransferase (HGSNAT; EC2.3.1.78) cause MPS IIIC (OMIM#252930), typified by impaired cognition, sleep–wake cycle changes, hyperactivity and early death, often before adulthood. The precise disease mechanism that causes symptom emergence remains unknown, posing a significant challenge in the development of effective therapeutics. As HGSNAT is conserved in Drosophila melanogaster, we now describe the creation and characterisation of the first Drosophila models of MPS IIIC. Flies with either an endogenous insertion mutation or RNAi-mediated knockdown of hgsnat were confirmed to have a reduced level of HGSNAT transcripts and age-dependent accumulation of heparan sulphate leading to engorgement of the endo/lysosomal compartment. This resulted in abnormalities at the pre-synapse, defective climbing and reduced overall activity. Altered circadian rhythms (shift in peak morning activity) were seen in hgsnat neuronal knockdown lines. Further, when hgsnat was knocked down in specific glial subsets (wrapping, cortical, astrocytes or subperineural glia), impaired climbing or reduced activity was noted, implying that hgsnat function in these specific glial subtypes contributes significantly to this behaviour and targeting treatments to these cell groups may be necessary to ameliorate or prevent symptom onset. These novel models of MPS IIIC provide critical research tools for delineating the key cellular pathways causal in the onset of neurodegeneration in this presently untreatable disorder.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
