Tao Liu, Zejun Peng, Manting Lai, Long Hu and Junfeng Zhao*,
{"title":"使用瞬时保护氨基酸进行反肽合成。","authors":"Tao Liu, Zejun Peng, Manting Lai, Long Hu and Junfeng Zhao*, ","doi":"10.1021/jacs.4c00314","DOIUrl":null,"url":null,"abstract":"<p >Peptide therapeutics have experienced a rapid resurgence over the past three decades. While a few peptide drugs are biologically produced, most are manufactured via chemical synthesis. The cycle of prior protection of the amino group of an α-amino acid, activation of its carboxyl group, aminolysis with the free amino group of a growing peptide chain, and deprotection of the N-terminus constitutes the principle of conventional C → N peptide chemical synthesis. The mandatory use of the N<i><sup>α</sup></i>-protecting group invokes two additional operations for incorporating each amino acid, resulting in poor step- and atom-economy. The burgeoning demand in the peptide therapeutic market necessitates cost-effective and environmentally friendly peptide manufacturing strategies. Inverse peptide chemical synthesis using unprotected amino acids has been proposed as an ideal and appealing strategy. However, it has remained unsuccessful for over 60 years due to severe racemization/epimerization during N → C peptide chain elongation. Herein, this challenge has been successfully addressed by ynamide coupling reagent employing a transient protection strategy. The activation, transient protection, aminolysis, and <i>in situ</i> deprotection were performed in one pot, thus offering a practical peptide chemical synthesis strategy formally using unprotected amino acids as the starting material. Its robustness was exemplified by syntheses of peptide active pharmaceutical ingredients. It is also amenable to fragment condensation and inverse solid-phase peptide synthesis. The compatibility to green solvents further enhances its application potential in large-scale peptide production. This study offered a cost-effective, operational convenient, and environmentally benign approach to peptides.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"146 6","pages":"4270–4280"},"PeriodicalIF":16.6000,"publicationDate":"2024-02-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Inverse Peptide Synthesis Using Transient Protected Amino Acids\",\"authors\":\"Tao Liu, Zejun Peng, Manting Lai, Long Hu and Junfeng Zhao*, \",\"doi\":\"10.1021/jacs.4c00314\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Peptide therapeutics have experienced a rapid resurgence over the past three decades. While a few peptide drugs are biologically produced, most are manufactured via chemical synthesis. The cycle of prior protection of the amino group of an α-amino acid, activation of its carboxyl group, aminolysis with the free amino group of a growing peptide chain, and deprotection of the N-terminus constitutes the principle of conventional C → N peptide chemical synthesis. The mandatory use of the N<i><sup>α</sup></i>-protecting group invokes two additional operations for incorporating each amino acid, resulting in poor step- and atom-economy. The burgeoning demand in the peptide therapeutic market necessitates cost-effective and environmentally friendly peptide manufacturing strategies. Inverse peptide chemical synthesis using unprotected amino acids has been proposed as an ideal and appealing strategy. However, it has remained unsuccessful for over 60 years due to severe racemization/epimerization during N → C peptide chain elongation. Herein, this challenge has been successfully addressed by ynamide coupling reagent employing a transient protection strategy. The activation, transient protection, aminolysis, and <i>in situ</i> deprotection were performed in one pot, thus offering a practical peptide chemical synthesis strategy formally using unprotected amino acids as the starting material. Its robustness was exemplified by syntheses of peptide active pharmaceutical ingredients. It is also amenable to fragment condensation and inverse solid-phase peptide synthesis. The compatibility to green solvents further enhances its application potential in large-scale peptide production. This study offered a cost-effective, operational convenient, and environmentally benign approach to peptides.</p>\",\"PeriodicalId\":49,\"journal\":{\"name\":\"Journal of the American Chemical Society\",\"volume\":\"146 6\",\"pages\":\"4270–4280\"},\"PeriodicalIF\":16.6000,\"publicationDate\":\"2024-02-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jacs.4c00314\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.4c00314","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
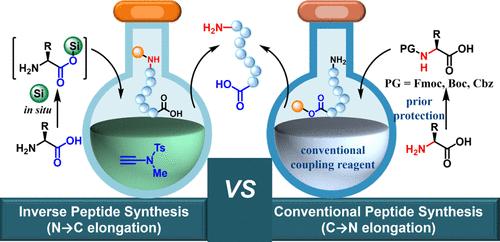
过去三十年来,多肽疗法迅速崛起。虽然有少数肽类药物是生物制剂,但大多数是通过化学合成制造的。传统 C → N 多肽化学合成的原理是事先保护α-氨基酸的氨基、活化α-氨基酸的羧基、与生长肽链的游离氨基发生氨解反应、去保护 N-末端。由于必须使用 Nα 保护基团,每个氨基酸的合成都要进行两次额外的操作,因此步骤和原子的经济性都很差。多肽治疗市场的需求急剧增长,这就需要成本效益高且环保的多肽生产策略。使用无保护氨基酸进行反肽化学合成被认为是一种理想而有吸引力的策略。然而,由于在 N → C 肽链伸长过程中存在严重的消旋化/二聚化现象,60 多年来这种方法一直未能成功。在此,采用瞬时保护策略的namide偶联试剂成功地解决了这一难题。活化、瞬时保护、氨基分解和原位脱保护在一锅内完成,从而提供了一种以未受保护的氨基酸为起始原料的实用多肽化学合成策略。多肽活性药物成分的合成证明了该方法的稳健性。它还适用于片段缩合和反向固相肽合成。与绿色溶剂的兼容性进一步提高了它在大规模多肽生产中的应用潜力。这项研究提供了一种成本效益高、操作方便、对环境无害的多肽方法。
Inverse Peptide Synthesis Using Transient Protected Amino Acids
Peptide therapeutics have experienced a rapid resurgence over the past three decades. While a few peptide drugs are biologically produced, most are manufactured via chemical synthesis. The cycle of prior protection of the amino group of an α-amino acid, activation of its carboxyl group, aminolysis with the free amino group of a growing peptide chain, and deprotection of the N-terminus constitutes the principle of conventional C → N peptide chemical synthesis. The mandatory use of the Nα-protecting group invokes two additional operations for incorporating each amino acid, resulting in poor step- and atom-economy. The burgeoning demand in the peptide therapeutic market necessitates cost-effective and environmentally friendly peptide manufacturing strategies. Inverse peptide chemical synthesis using unprotected amino acids has been proposed as an ideal and appealing strategy. However, it has remained unsuccessful for over 60 years due to severe racemization/epimerization during N → C peptide chain elongation. Herein, this challenge has been successfully addressed by ynamide coupling reagent employing a transient protection strategy. The activation, transient protection, aminolysis, and in situ deprotection were performed in one pot, thus offering a practical peptide chemical synthesis strategy formally using unprotected amino acids as the starting material. Its robustness was exemplified by syntheses of peptide active pharmaceutical ingredients. It is also amenable to fragment condensation and inverse solid-phase peptide synthesis. The compatibility to green solvents further enhances its application potential in large-scale peptide production. This study offered a cost-effective, operational convenient, and environmentally benign approach to peptides.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
