Joke Deschildre, Boris Vandemoortele, Jens Uwe Loers, Katleen De Preter, Vanessa Vermeirssen
{"title":"评估精准肿瘤学的单样本网络推断方法。","authors":"Joke Deschildre, Boris Vandemoortele, Jens Uwe Loers, Katleen De Preter, Vanessa Vermeirssen","doi":"10.1038/s41540-024-00340-w","DOIUrl":null,"url":null,"abstract":"<p><p>A major challenge in precision oncology is to detect targetable cancer vulnerabilities in individual patients. Modeling high-throughput omics data in biological networks allows identifying key molecules and processes of tumorigenesis. Traditionally, network inference methods rely on many samples to contain sufficient information for learning, resulting in aggregate networks. However, to implement patient-tailored approaches in precision oncology, we need to interpret omics data at the level of individual patients. Several single-sample network inference methods have been developed that infer biological networks for an individual sample from bulk RNA-seq data. However, only a limited comparison of these methods has been made and many methods rely on 'normal tissue' samples as reference, which are not always available. Here, we conducted an evaluation of the single-sample network inference methods SSN, LIONESS, SWEET, iENA, CSN and SSPGI using transcriptomic profiles of lung and brain cancer cell lines from the CCLE database. The methods constructed functional gene networks with distinct network characteristics. Hub gene analyses revealed different degrees of subtype-specificity across methods. Single-sample networks were able to distinguish between tumor subtypes, as exemplified by node strength clustering, enrichment of known subtype-specific driver genes among hubs and differential node strength. We also showed that single-sample networks correlated better to other omics data from the same cell line as compared to aggregate networks. We conclude that single-sample network inference methods can reflect sample-specific biology when 'normal tissue' samples are absent and we point out peculiarities of each method.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"10 1","pages":"18"},"PeriodicalIF":4.4000,"publicationDate":"2024-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10869342/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evaluation of single-sample network inference methods for precision oncology.\",\"authors\":\"Joke Deschildre, Boris Vandemoortele, Jens Uwe Loers, Katleen De Preter, Vanessa Vermeirssen\",\"doi\":\"10.1038/s41540-024-00340-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A major challenge in precision oncology is to detect targetable cancer vulnerabilities in individual patients. Modeling high-throughput omics data in biological networks allows identifying key molecules and processes of tumorigenesis. Traditionally, network inference methods rely on many samples to contain sufficient information for learning, resulting in aggregate networks. However, to implement patient-tailored approaches in precision oncology, we need to interpret omics data at the level of individual patients. Several single-sample network inference methods have been developed that infer biological networks for an individual sample from bulk RNA-seq data. However, only a limited comparison of these methods has been made and many methods rely on 'normal tissue' samples as reference, which are not always available. Here, we conducted an evaluation of the single-sample network inference methods SSN, LIONESS, SWEET, iENA, CSN and SSPGI using transcriptomic profiles of lung and brain cancer cell lines from the CCLE database. The methods constructed functional gene networks with distinct network characteristics. Hub gene analyses revealed different degrees of subtype-specificity across methods. Single-sample networks were able to distinguish between tumor subtypes, as exemplified by node strength clustering, enrichment of known subtype-specific driver genes among hubs and differential node strength. We also showed that single-sample networks correlated better to other omics data from the same cell line as compared to aggregate networks. We conclude that single-sample network inference methods can reflect sample-specific biology when 'normal tissue' samples are absent and we point out peculiarities of each method.</p>\",\"PeriodicalId\":19345,\"journal\":{\"name\":\"NPJ Systems Biology and Applications\",\"volume\":\"10 1\",\"pages\":\"18\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2024-02-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10869342/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Systems Biology and Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41540-024-00340-w\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00340-w","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Evaluation of single-sample network inference methods for precision oncology.
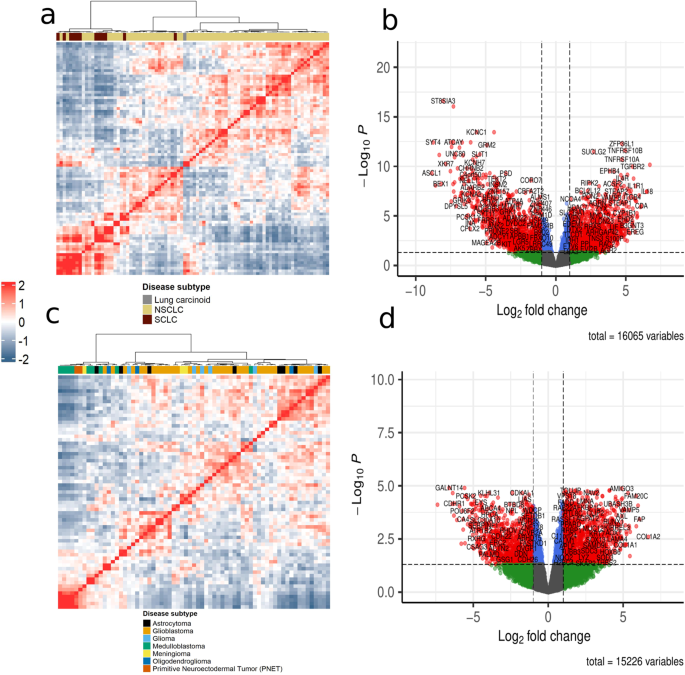
A major challenge in precision oncology is to detect targetable cancer vulnerabilities in individual patients. Modeling high-throughput omics data in biological networks allows identifying key molecules and processes of tumorigenesis. Traditionally, network inference methods rely on many samples to contain sufficient information for learning, resulting in aggregate networks. However, to implement patient-tailored approaches in precision oncology, we need to interpret omics data at the level of individual patients. Several single-sample network inference methods have been developed that infer biological networks for an individual sample from bulk RNA-seq data. However, only a limited comparison of these methods has been made and many methods rely on 'normal tissue' samples as reference, which are not always available. Here, we conducted an evaluation of the single-sample network inference methods SSN, LIONESS, SWEET, iENA, CSN and SSPGI using transcriptomic profiles of lung and brain cancer cell lines from the CCLE database. The methods constructed functional gene networks with distinct network characteristics. Hub gene analyses revealed different degrees of subtype-specificity across methods. Single-sample networks were able to distinguish between tumor subtypes, as exemplified by node strength clustering, enrichment of known subtype-specific driver genes among hubs and differential node strength. We also showed that single-sample networks correlated better to other omics data from the same cell line as compared to aggregate networks. We conclude that single-sample network inference methods can reflect sample-specific biology when 'normal tissue' samples are absent and we point out peculiarities of each method.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
