{"title":"患有史密斯-马盖尼斯样综合征的 CHD2 兄弟姐妹中的低度父母性腺嵌合。","authors":"Francesca Cogliati, Letizia Straniero, Valeria Rimoldi, Maura Masciadri, Sara Perego, Berardo Rinaldi, Donatella Milani, Davide Gentilini, Lidia Larizza, Rosanna Asselta, Silvia Russo, Maria Francesca Bedeschi","doi":"10.1002/ajmg.b.32976","DOIUrl":null,"url":null,"abstract":"<p>Loss-of-function <i>CHD2</i> (chromodomain helicase DNA-binding protein 2) mutations are associated with a spectrum of neurodevelopmental disorders often including early-onset generalized seizures, photosensitivity, and epileptic encephalopathies. Patients show psychomotor delay/intellectual disability (ID), autistic features, and behavior disorders, such as aggression and impulsivity. Most reported cases are sporadic with description of germline mosaicism only in two families. We detect the first case of parental gonosomal <i>CHD2</i> mosaicism disclosed by two brothers showing mild ID, born to healthy parents. The eldest brother has a history of drug-controlled generalized tonic–clonic seizures and displays sleep disorder and aggressive behavior suggestive of Smith–Magenis syndrome (SMS). Analysis of brothers’ DNAs by next-generation sequencing (NGS) custom gene panel for pediatric epilepsy and/or ID disclosed in both the same pathogenic <i>CHD2</i> variant. Additional NGS experiment on genomic DNA from parents’ peripheral blood and from buccal swab raised the suspicion of low-grade gonosomal mosaicism in the unaffected mother subsequently confirmed by digital polymerase chain reaction (dPCR). This report underlines as worthwhile <i>CHD2</i> screening in individuals presenting ID/developmental delay, with/without epilepsy, and behavior and sleep disorders suggestive of SMS. Detecting a <i>CHD2</i> variant should prime testing probands' parents by NGS coupled to dPCR on different tissues to exclude/confirm gonosomal mosaicism and define the recurrence risk.</p>","PeriodicalId":7673,"journal":{"name":"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics","volume":"195 6","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2024-02-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.b.32976","citationCount":"0","resultStr":"{\"title\":\"Low-grade parental gonosomal mosaicism in CHD2 siblings with Smith–Magenis-like syndrome\",\"authors\":\"Francesca Cogliati, Letizia Straniero, Valeria Rimoldi, Maura Masciadri, Sara Perego, Berardo Rinaldi, Donatella Milani, Davide Gentilini, Lidia Larizza, Rosanna Asselta, Silvia Russo, Maria Francesca Bedeschi\",\"doi\":\"10.1002/ajmg.b.32976\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Loss-of-function <i>CHD2</i> (chromodomain helicase DNA-binding protein 2) mutations are associated with a spectrum of neurodevelopmental disorders often including early-onset generalized seizures, photosensitivity, and epileptic encephalopathies. Patients show psychomotor delay/intellectual disability (ID), autistic features, and behavior disorders, such as aggression and impulsivity. Most reported cases are sporadic with description of germline mosaicism only in two families. We detect the first case of parental gonosomal <i>CHD2</i> mosaicism disclosed by two brothers showing mild ID, born to healthy parents. The eldest brother has a history of drug-controlled generalized tonic–clonic seizures and displays sleep disorder and aggressive behavior suggestive of Smith–Magenis syndrome (SMS). Analysis of brothers’ DNAs by next-generation sequencing (NGS) custom gene panel for pediatric epilepsy and/or ID disclosed in both the same pathogenic <i>CHD2</i> variant. Additional NGS experiment on genomic DNA from parents’ peripheral blood and from buccal swab raised the suspicion of low-grade gonosomal mosaicism in the unaffected mother subsequently confirmed by digital polymerase chain reaction (dPCR). This report underlines as worthwhile <i>CHD2</i> screening in individuals presenting ID/developmental delay, with/without epilepsy, and behavior and sleep disorders suggestive of SMS. Detecting a <i>CHD2</i> variant should prime testing probands' parents by NGS coupled to dPCR on different tissues to exclude/confirm gonosomal mosaicism and define the recurrence risk.</p>\",\"PeriodicalId\":7673,\"journal\":{\"name\":\"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics\",\"volume\":\"195 6\",\"pages\":\"\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-02-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.b.32976\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.b.32976\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.b.32976","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
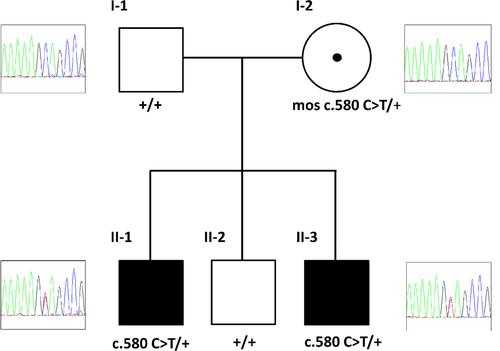
功能缺失型 CHD2(染色质链螺旋酶 DNA 结合蛋白 2)突变与一系列神经发育障碍有关,通常包括早发型全身性癫痫发作、光敏感性和癫痫性脑病。患者表现出精神运动发育迟缓/智力障碍(ID)、自闭症特征和行为障碍,如攻击性和冲动性。大多数报道的病例都是散发性的,只有两个家族描述了种系镶嵌现象。我们发现了首例父母性染色体 CHD2 嵌合的病例,两兄弟表现为轻度 ID,父母均健康。长兄有药物控制的全身强直-阵挛发作病史,并表现出睡眠障碍和攻击性行为,提示患有史密斯-马吉尼斯综合征(SMS)。通过下一代测序(NGS)定制的小儿癫痫和/或 ID 基因面板对兄弟俩的 DNA 进行分析,发现他们都有相同的致病性 CHD2 变异。对来自父母外周血和口腔拭子的基因组 DNA 进行的其他 NGS 实验,使人们怀疑未受影响的母亲体内存在低度性染色体嵌合,随后通过数字聚合酶链反应(dPCR)证实了这一点。该报告强调,在出现 ID/发育迟缓、伴有/不伴有癫痫以及行为和睡眠障碍并提示 SMS 的个体中,CHD2 筛查是值得的。检测 CHD2 变异时,应首先通过 NGS 和 dPCR 对不同组织进行检测,以排除/确认染色体嵌合,并确定复发风险。
Low-grade parental gonosomal mosaicism in CHD2 siblings with Smith–Magenis-like syndrome
Loss-of-function CHD2 (chromodomain helicase DNA-binding protein 2) mutations are associated with a spectrum of neurodevelopmental disorders often including early-onset generalized seizures, photosensitivity, and epileptic encephalopathies. Patients show psychomotor delay/intellectual disability (ID), autistic features, and behavior disorders, such as aggression and impulsivity. Most reported cases are sporadic with description of germline mosaicism only in two families. We detect the first case of parental gonosomal CHD2 mosaicism disclosed by two brothers showing mild ID, born to healthy parents. The eldest brother has a history of drug-controlled generalized tonic–clonic seizures and displays sleep disorder and aggressive behavior suggestive of Smith–Magenis syndrome (SMS). Analysis of brothers’ DNAs by next-generation sequencing (NGS) custom gene panel for pediatric epilepsy and/or ID disclosed in both the same pathogenic CHD2 variant. Additional NGS experiment on genomic DNA from parents’ peripheral blood and from buccal swab raised the suspicion of low-grade gonosomal mosaicism in the unaffected mother subsequently confirmed by digital polymerase chain reaction (dPCR). This report underlines as worthwhile CHD2 screening in individuals presenting ID/developmental delay, with/without epilepsy, and behavior and sleep disorders suggestive of SMS. Detecting a CHD2 variant should prime testing probands' parents by NGS coupled to dPCR on different tissues to exclude/confirm gonosomal mosaicism and define the recurrence risk.
期刊介绍:
Neuropsychiatric Genetics, Part B of the American Journal of Medical Genetics (AJMG) , provides a forum for experimental and clinical investigations of the genetic mechanisms underlying neurologic and psychiatric disorders. It is a resource for novel genetics studies of the heritable nature of psychiatric and other nervous system disorders, characterized at the molecular, cellular or behavior levels. Neuropsychiatric Genetics publishes eight times per year.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
