Gavin Esson, Ian Logan, Katrina Wood, Andrew C Browning, John A Sayer
{"title":"肾衰竭患者中与 NPHP1 同源全基因缺失相关的多种视网膜-肾脏表型。","authors":"Gavin Esson, Ian Logan, Katrina Wood, Andrew C Browning, John A Sayer","doi":"10.1007/s44162-024-00031-4","DOIUrl":null,"url":null,"abstract":"<p><p>A precise diagnosis in medicine allows appropriate disease-specific management. Kidney failure of unknown aetiology remains a frequent diagnostic label within the haemodialysis unit and kidney transplant clinic, accounting for 15-20% of these patients. Approximately 10% of such cases may have an underlying monogenic cause of kidney failure. Modern genetic approaches can provide a precise diagnosis for patients and their families. A search for extra-renal disease manifestations is also important as this may point to a specific genetic diagnosis. Here, we present two patients where molecular genetic testing was performed because of kidney failure of unknown aetiology and associated retinal phenotypes. The first patient reached kidney failure at 16 years of age but only presented with a retinal phenotype at 59 years of age and was found to have evidence of rod-cone dystrophy. The second patient presented with childhood kidney failure at the age of 15 years and developed visual difficulties and photophobia at the age of 32 years and was diagnosed with cone dystrophy. In both cases, genetic tests were performed which revealed a homozygous whole-gene deletion of <i>NPHP1</i>-encoding nephrocystin-1, providing the unifying diagnosis of Senior-Løken syndrome type 1. We conclude that reviewing kidney and extra-renal phenotypes together with targeted genetic testing was informative in these cases of kidney failure of unknown aetiology and associated retinal phenotypes. The involvement of an interdisciplinary team is advisable when managing such patients and allows referral to other relevant specialities. The long time lag and lack of diagnostic clarity and clinical evaluation in our cases should encourage genetic investigations for every young patient with unexplained kidney failure. For these and similar patients, a more timely genetic diagnosis would allow for improved management, a risk assessment of kidney disease in relatives, and the earlier identification of extra-renal disease manifestations.</p><p><strong>Supplementary information: </strong>The online version contains supplementary material available at 10.1007/s44162-024-00031-4.</p>","PeriodicalId":73925,"journal":{"name":"Journal of rare diseases (Berlin, Germany)","volume":"3 1","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10904492/pdf/","citationCount":"0","resultStr":"{\"title\":\"Diverse retinal-kidney phenotypes associated with <i>NPHP1</i> homozygous whole-gene deletions in patients with kidney failure.\",\"authors\":\"Gavin Esson, Ian Logan, Katrina Wood, Andrew C Browning, John A Sayer\",\"doi\":\"10.1007/s44162-024-00031-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A precise diagnosis in medicine allows appropriate disease-specific management. Kidney failure of unknown aetiology remains a frequent diagnostic label within the haemodialysis unit and kidney transplant clinic, accounting for 15-20% of these patients. Approximately 10% of such cases may have an underlying monogenic cause of kidney failure. Modern genetic approaches can provide a precise diagnosis for patients and their families. A search for extra-renal disease manifestations is also important as this may point to a specific genetic diagnosis. Here, we present two patients where molecular genetic testing was performed because of kidney failure of unknown aetiology and associated retinal phenotypes. The first patient reached kidney failure at 16 years of age but only presented with a retinal phenotype at 59 years of age and was found to have evidence of rod-cone dystrophy. The second patient presented with childhood kidney failure at the age of 15 years and developed visual difficulties and photophobia at the age of 32 years and was diagnosed with cone dystrophy. In both cases, genetic tests were performed which revealed a homozygous whole-gene deletion of <i>NPHP1</i>-encoding nephrocystin-1, providing the unifying diagnosis of Senior-Løken syndrome type 1. We conclude that reviewing kidney and extra-renal phenotypes together with targeted genetic testing was informative in these cases of kidney failure of unknown aetiology and associated retinal phenotypes. The involvement of an interdisciplinary team is advisable when managing such patients and allows referral to other relevant specialities. The long time lag and lack of diagnostic clarity and clinical evaluation in our cases should encourage genetic investigations for every young patient with unexplained kidney failure. For these and similar patients, a more timely genetic diagnosis would allow for improved management, a risk assessment of kidney disease in relatives, and the earlier identification of extra-renal disease manifestations.</p><p><strong>Supplementary information: </strong>The online version contains supplementary material available at 10.1007/s44162-024-00031-4.</p>\",\"PeriodicalId\":73925,\"journal\":{\"name\":\"Journal of rare diseases (Berlin, Germany)\",\"volume\":\"3 1\",\"pages\":\"7\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10904492/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of rare diseases (Berlin, Germany)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s44162-024-00031-4\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of rare diseases (Berlin, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s44162-024-00031-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/1 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Diverse retinal-kidney phenotypes associated with NPHP1 homozygous whole-gene deletions in patients with kidney failure.
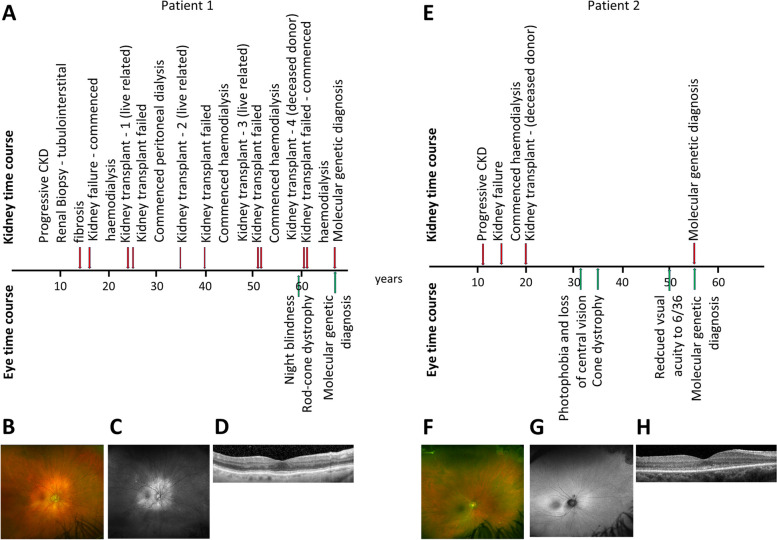
A precise diagnosis in medicine allows appropriate disease-specific management. Kidney failure of unknown aetiology remains a frequent diagnostic label within the haemodialysis unit and kidney transplant clinic, accounting for 15-20% of these patients. Approximately 10% of such cases may have an underlying monogenic cause of kidney failure. Modern genetic approaches can provide a precise diagnosis for patients and their families. A search for extra-renal disease manifestations is also important as this may point to a specific genetic diagnosis. Here, we present two patients where molecular genetic testing was performed because of kidney failure of unknown aetiology and associated retinal phenotypes. The first patient reached kidney failure at 16 years of age but only presented with a retinal phenotype at 59 years of age and was found to have evidence of rod-cone dystrophy. The second patient presented with childhood kidney failure at the age of 15 years and developed visual difficulties and photophobia at the age of 32 years and was diagnosed with cone dystrophy. In both cases, genetic tests were performed which revealed a homozygous whole-gene deletion of NPHP1-encoding nephrocystin-1, providing the unifying diagnosis of Senior-Løken syndrome type 1. We conclude that reviewing kidney and extra-renal phenotypes together with targeted genetic testing was informative in these cases of kidney failure of unknown aetiology and associated retinal phenotypes. The involvement of an interdisciplinary team is advisable when managing such patients and allows referral to other relevant specialities. The long time lag and lack of diagnostic clarity and clinical evaluation in our cases should encourage genetic investigations for every young patient with unexplained kidney failure. For these and similar patients, a more timely genetic diagnosis would allow for improved management, a risk assessment of kidney disease in relatives, and the earlier identification of extra-renal disease manifestations.
Supplementary information: The online version contains supplementary material available at 10.1007/s44162-024-00031-4.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
