Pauline J. Ollitrault*, Matthias Loipersberger, Robert M. Parrish, Alexander Erhard, Christine Maier, Christian Sommer, Juris Ulmanis, Thomas Monz, Christian Gogolin, Christofer S. Tautermann, Gian-Luca R. Anselmetti, Matthias Degroote, Nikolaj Moll, Raffaele Santagati and Michael Streif*,
{"title":"在陷波离子量子计算机上估算静电相互作用能量","authors":"Pauline J. Ollitrault*, Matthias Loipersberger, Robert M. Parrish, Alexander Erhard, Christine Maier, Christian Sommer, Juris Ulmanis, Thomas Monz, Christian Gogolin, Christofer S. Tautermann, Gian-Luca R. Anselmetti, Matthias Degroote, Nikolaj Moll, Raffaele Santagati and Michael Streif*, ","doi":"10.1021/acscentsci.4c00058","DOIUrl":null,"url":null,"abstract":"<p >We present the first hardware implementation of electrostatic interaction energies by using a trapped-ion quantum computer. As test system for our computation, we focus on the reduction of NO to N<sub>2</sub>O catalyzed by a nitric oxide reductase (NOR). The quantum computer is used to generate an approximate ground state within the NOR active space. To efficiently measure the necessary one-particle density matrices, we incorporate fermionic basis rotations into the quantum circuit without extending the circuit length, laying the groundwork for further efficient measurement routines using factorizations. Measurements in the computational basis are then used as inputs for computing the electrostatic interaction energies on a classical computer. Our experimental results strongly agree with classical noise-less simulations of the same circuits, finding electrostatic interaction energies within chemical accuracy despite hardware noise. This work shows that algorithms tailored to specific observables of interest, such as interaction energies, may require significantly fewer quantum resources than individual ground state energies would require in the straightforward supermolecular approach.</p><p >The first demonstration of the computation of electrostatic interaction energies on a trapped-ion quantum computer, tested on the reduction of NO to N<sub>2</sub>O, shows strong agreement with classical simulations.</p>","PeriodicalId":10,"journal":{"name":"ACS Central Science","volume":null,"pages":null},"PeriodicalIF":12.7000,"publicationDate":"2024-03-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acscentsci.4c00058","citationCount":"0","resultStr":"{\"title\":\"Estimation of Electrostatic Interaction Energies on a Trapped-Ion Quantum Computer\",\"authors\":\"Pauline J. Ollitrault*, Matthias Loipersberger, Robert M. Parrish, Alexander Erhard, Christine Maier, Christian Sommer, Juris Ulmanis, Thomas Monz, Christian Gogolin, Christofer S. Tautermann, Gian-Luca R. Anselmetti, Matthias Degroote, Nikolaj Moll, Raffaele Santagati and Michael Streif*, \",\"doi\":\"10.1021/acscentsci.4c00058\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present the first hardware implementation of electrostatic interaction energies by using a trapped-ion quantum computer. As test system for our computation, we focus on the reduction of NO to N<sub>2</sub>O catalyzed by a nitric oxide reductase (NOR). The quantum computer is used to generate an approximate ground state within the NOR active space. To efficiently measure the necessary one-particle density matrices, we incorporate fermionic basis rotations into the quantum circuit without extending the circuit length, laying the groundwork for further efficient measurement routines using factorizations. Measurements in the computational basis are then used as inputs for computing the electrostatic interaction energies on a classical computer. Our experimental results strongly agree with classical noise-less simulations of the same circuits, finding electrostatic interaction energies within chemical accuracy despite hardware noise. This work shows that algorithms tailored to specific observables of interest, such as interaction energies, may require significantly fewer quantum resources than individual ground state energies would require in the straightforward supermolecular approach.</p><p >The first demonstration of the computation of electrostatic interaction energies on a trapped-ion quantum computer, tested on the reduction of NO to N<sub>2</sub>O, shows strong agreement with classical simulations.</p>\",\"PeriodicalId\":10,\"journal\":{\"name\":\"ACS Central Science\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":12.7000,\"publicationDate\":\"2024-03-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acscentsci.4c00058\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Central Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscentsci.4c00058\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Central Science","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscentsci.4c00058","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
我们首次利用困离子量子计算机对静电相互作用能量进行了硬件实现。作为计算的测试系统,我们重点研究了一氧化氮还原酶(NOR)催化 NO 还原成 N2O 的过程。量子计算机用于生成 NOR 活性空间内的近似基态。为了有效测量必要的单粒子密度矩阵,我们在不延长电路长度的情况下将费米子基旋转纳入量子电路,为进一步使用因式分解进行高效测量奠定了基础。然后,计算基础中的测量结果将作为在经典计算机上计算静电相互作用能量的输入。我们的实验结果与相同电路的经典无噪声仿真结果非常吻合,尽管存在硬件噪声,但找到的静电相互作用能仍在化学精度范围内。这项工作表明,与直接的超分子方法计算单个基态能量所需的量子资源相比,针对相互作用能量等特定观测指标量身定制的算法所需的量子资源可能要少得多。
Estimation of Electrostatic Interaction Energies on a Trapped-Ion Quantum Computer
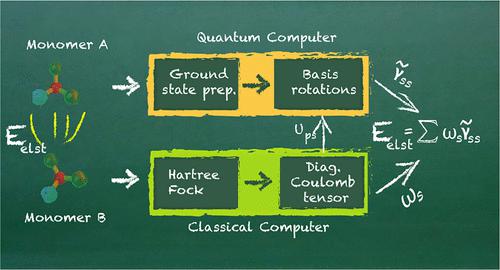
We present the first hardware implementation of electrostatic interaction energies by using a trapped-ion quantum computer. As test system for our computation, we focus on the reduction of NO to N2O catalyzed by a nitric oxide reductase (NOR). The quantum computer is used to generate an approximate ground state within the NOR active space. To efficiently measure the necessary one-particle density matrices, we incorporate fermionic basis rotations into the quantum circuit without extending the circuit length, laying the groundwork for further efficient measurement routines using factorizations. Measurements in the computational basis are then used as inputs for computing the electrostatic interaction energies on a classical computer. Our experimental results strongly agree with classical noise-less simulations of the same circuits, finding electrostatic interaction energies within chemical accuracy despite hardware noise. This work shows that algorithms tailored to specific observables of interest, such as interaction energies, may require significantly fewer quantum resources than individual ground state energies would require in the straightforward supermolecular approach.
The first demonstration of the computation of electrostatic interaction energies on a trapped-ion quantum computer, tested on the reduction of NO to N2O, shows strong agreement with classical simulations.
期刊介绍:
ACS Central Science publishes significant primary reports on research in chemistry and allied fields where chemical approaches are pivotal. As the first fully open-access journal by the American Chemical Society, it covers compelling and important contributions to the broad chemistry and scientific community. "Central science," a term popularized nearly 40 years ago, emphasizes chemistry's central role in connecting physical and life sciences, and fundamental sciences with applied disciplines like medicine and engineering. The journal focuses on exceptional quality articles, addressing advances in fundamental chemistry and interdisciplinary research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
