Ataur Katebi, Xiaowen Chen, Daniel Ramirez, Sheng Li, Mingyang Lu
{"title":"数据驱动的 IDH 突变型急性髓细胞白血病发生的核心基因调控网络建模","authors":"Ataur Katebi, Xiaowen Chen, Daniel Ramirez, Sheng Li, Mingyang Lu","doi":"10.1038/s41540-024-00366-0","DOIUrl":null,"url":null,"abstract":"<p>Acute myeloid leukemia (AML) is characterized by uncontrolled proliferation of poorly differentiated myeloid cells, with a heterogenous mutational landscape. Mutations in IDH1 and IDH2 are found in 20% of the AML cases. Although much effort has been made to identify genes associated with leukemogenesis, the regulatory mechanism of AML state transition is still not fully understood. To alleviate this issue, here we develop a new computational approach that integrates genomic data from diverse sources, including gene expression and ATAC-seq datasets, curated gene regulatory interaction databases, and mathematical modeling to establish models of context-specific core gene regulatory networks (GRNs) for a mechanistic understanding of tumorigenesis of AML with IDH mutations. The approach adopts a new optimization procedure to identify the top network according to its accuracy in capturing gene expression states and its flexibility to allow sufficient control of state transitions. From GRN modeling, we identify key regulators associated with the function of IDH mutations, such as DNA methyltransferase DNMT1, and network destabilizers, such as E2F1. The constructed core regulatory network and outcomes of in-silico network perturbations are supported by survival data from AML patients. We expect that the combined bioinformatics and systems-biology modeling approach will be generally applicable to elucidate the gene regulation of disease progression.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"121 1","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2024-04-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Data-driven modeling of core gene regulatory network underlying leukemogenesis in IDH mutant AML\",\"authors\":\"Ataur Katebi, Xiaowen Chen, Daniel Ramirez, Sheng Li, Mingyang Lu\",\"doi\":\"10.1038/s41540-024-00366-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Acute myeloid leukemia (AML) is characterized by uncontrolled proliferation of poorly differentiated myeloid cells, with a heterogenous mutational landscape. Mutations in IDH1 and IDH2 are found in 20% of the AML cases. Although much effort has been made to identify genes associated with leukemogenesis, the regulatory mechanism of AML state transition is still not fully understood. To alleviate this issue, here we develop a new computational approach that integrates genomic data from diverse sources, including gene expression and ATAC-seq datasets, curated gene regulatory interaction databases, and mathematical modeling to establish models of context-specific core gene regulatory networks (GRNs) for a mechanistic understanding of tumorigenesis of AML with IDH mutations. The approach adopts a new optimization procedure to identify the top network according to its accuracy in capturing gene expression states and its flexibility to allow sufficient control of state transitions. From GRN modeling, we identify key regulators associated with the function of IDH mutations, such as DNA methyltransferase DNMT1, and network destabilizers, such as E2F1. The constructed core regulatory network and outcomes of in-silico network perturbations are supported by survival data from AML patients. We expect that the combined bioinformatics and systems-biology modeling approach will be generally applicable to elucidate the gene regulation of disease progression.</p>\",\"PeriodicalId\":19345,\"journal\":{\"name\":\"NPJ Systems Biology and Applications\",\"volume\":\"121 1\",\"pages\":\"\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-04-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Systems Biology and Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41540-024-00366-0\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00366-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Data-driven modeling of core gene regulatory network underlying leukemogenesis in IDH mutant AML
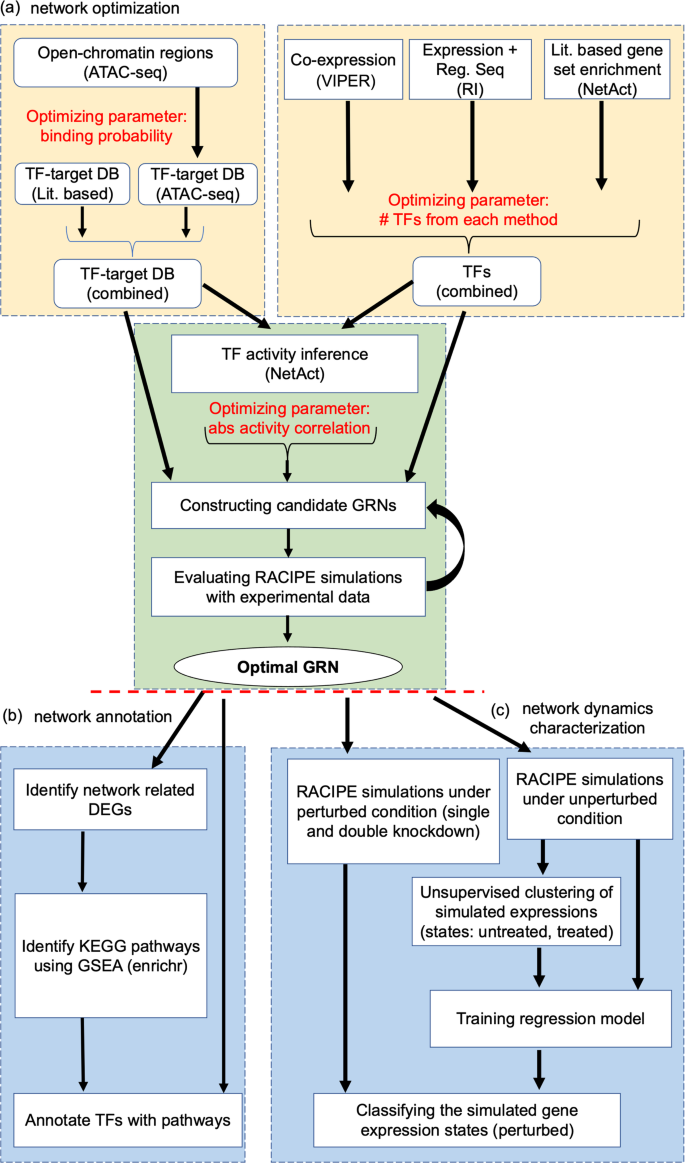
Acute myeloid leukemia (AML) is characterized by uncontrolled proliferation of poorly differentiated myeloid cells, with a heterogenous mutational landscape. Mutations in IDH1 and IDH2 are found in 20% of the AML cases. Although much effort has been made to identify genes associated with leukemogenesis, the regulatory mechanism of AML state transition is still not fully understood. To alleviate this issue, here we develop a new computational approach that integrates genomic data from diverse sources, including gene expression and ATAC-seq datasets, curated gene regulatory interaction databases, and mathematical modeling to establish models of context-specific core gene regulatory networks (GRNs) for a mechanistic understanding of tumorigenesis of AML with IDH mutations. The approach adopts a new optimization procedure to identify the top network according to its accuracy in capturing gene expression states and its flexibility to allow sufficient control of state transitions. From GRN modeling, we identify key regulators associated with the function of IDH mutations, such as DNA methyltransferase DNMT1, and network destabilizers, such as E2F1. The constructed core regulatory network and outcomes of in-silico network perturbations are supported by survival data from AML patients. We expect that the combined bioinformatics and systems-biology modeling approach will be generally applicable to elucidate the gene regulation of disease progression.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
