Hindol Chatterjee , Anshuman J. Mahapatra , Martin Zacharias , Neelanjana Sengupta
{"title":"膜蛋白折叠过程中的螺旋重组:通过模拟细菌眼色素(BR)片段获得的启示。","authors":"Hindol Chatterjee , Anshuman J. Mahapatra , Martin Zacharias , Neelanjana Sengupta","doi":"10.1016/j.bbamem.2024.184333","DOIUrl":null,"url":null,"abstract":"<div><p>Membrane protein folding is distinct from folding of soluble proteins. Conformational acquisition in major membrane protein subclasses can be delineated into insertion and folding processes. An exception to the “two stage” folding, later developed to “three stage” folding, is observed within the last two helices in bacteriorhodopsin (BR), a system that serves as a model membrane protein. We employ a reductionist approach to understand interplay of molecular factors underlying the apparent defiance. Leveraging available solution NMR structures, we construct, sample in silico, and analyze partially (PIn) and fully inserted (FIn) BR membrane states. The membrane lateral C-terminal helix (CH) in PIn is markedly prone to transient structural distortions over microsecond timescales; a disorder prone region (DPR) is thereby identified. While clear transmembrane propensities are not acquired, the distortions induce alterations in local membrane curvature and area per lipid. Importantly, energetic decompositions reveal that overall, the N-terminal helix (NH) is thermodynamically more stable in the PIn. Higher overall stability of the FIn arises from favorable interactions between the NH and the CH. Our results establish lack of spontaneous transition of the PIn to the FIn, and attributes their partitioning to barriers that exceed those accessible with thermal fluctuations. This work paves the way for further detailed studies aimed at determining the thermo-kinetic roles of the initial five helices, or complementary external factors, in complete helical folding and insertion in BR. We comment that complementing such efforts with the growing field of machine learning assisted energy landscape searches may offer unprecedented insights.</p></div>","PeriodicalId":8831,"journal":{"name":"Biochimica et biophysica acta. Biomembranes","volume":"1866 5","pages":"Article 184333"},"PeriodicalIF":2.5000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Helical reorganization in the context of membrane protein folding: Insights from simulations with bacteriorhodopsin (BR) fragments\",\"authors\":\"Hindol Chatterjee , Anshuman J. Mahapatra , Martin Zacharias , Neelanjana Sengupta\",\"doi\":\"10.1016/j.bbamem.2024.184333\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Membrane protein folding is distinct from folding of soluble proteins. Conformational acquisition in major membrane protein subclasses can be delineated into insertion and folding processes. An exception to the “two stage” folding, later developed to “three stage” folding, is observed within the last two helices in bacteriorhodopsin (BR), a system that serves as a model membrane protein. We employ a reductionist approach to understand interplay of molecular factors underlying the apparent defiance. Leveraging available solution NMR structures, we construct, sample in silico, and analyze partially (PIn) and fully inserted (FIn) BR membrane states. The membrane lateral C-terminal helix (CH) in PIn is markedly prone to transient structural distortions over microsecond timescales; a disorder prone region (DPR) is thereby identified. While clear transmembrane propensities are not acquired, the distortions induce alterations in local membrane curvature and area per lipid. Importantly, energetic decompositions reveal that overall, the N-terminal helix (NH) is thermodynamically more stable in the PIn. Higher overall stability of the FIn arises from favorable interactions between the NH and the CH. Our results establish lack of spontaneous transition of the PIn to the FIn, and attributes their partitioning to barriers that exceed those accessible with thermal fluctuations. This work paves the way for further detailed studies aimed at determining the thermo-kinetic roles of the initial five helices, or complementary external factors, in complete helical folding and insertion in BR. We comment that complementing such efforts with the growing field of machine learning assisted energy landscape searches may offer unprecedented insights.</p></div>\",\"PeriodicalId\":8831,\"journal\":{\"name\":\"Biochimica et biophysica acta. Biomembranes\",\"volume\":\"1866 5\",\"pages\":\"Article 184333\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biochimica et biophysica acta. Biomembranes\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0005273624000646\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/5/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochimica et biophysica acta. Biomembranes","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0005273624000646","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/11 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
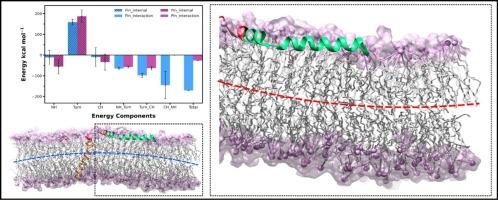
膜蛋白折叠不同于可溶性蛋白的折叠。主要膜蛋白亚类的构象获得可分为插入和折叠过程。在作为膜蛋白模型的细菌眼色素(BR)系统中,观察到了 "两阶段 "折叠(后来发展为 "三阶段 "折叠)的例外情况。我们采用还原论的方法来了解导致这种明显违抗的分子因素之间的相互作用。利用现有的溶液核磁共振结构,我们构建了部分(PIn)和完全插入(FIn)BR 膜状态,并对其进行了硅学取样和分析。在 PIn 中,膜侧 C 端螺旋(CH)在微秒时间尺度上明显容易发生瞬时结构扭曲;由此确定了一个易紊乱区域(DPR)。虽然没有获得明确的跨膜倾向性,但这种扭曲会引起局部膜曲率和单位脂质面积的改变。重要的是,能量分解显示,总体而言,N 端螺旋(NH)在 PIn 中的热力学稳定性更高。NH 与 CH 之间的有利相互作用使 FIn 整体稳定性更高。我们的研究结果表明,PIn 缺乏向 FIn 的自发转变,并将它们的分离归因于超过热波动所能达到的壁垒。这项工作为进一步详细研究铺平了道路,这些研究旨在确定最初五个螺旋或互补外部因素在完全螺旋折叠和插入 BR 中的热动力学作用。我们认为,将这些工作与不断发展的机器学习辅助能量景观搜索领域相辅相成,可能会提供前所未有的见解。
Helical reorganization in the context of membrane protein folding: Insights from simulations with bacteriorhodopsin (BR) fragments
Membrane protein folding is distinct from folding of soluble proteins. Conformational acquisition in major membrane protein subclasses can be delineated into insertion and folding processes. An exception to the “two stage” folding, later developed to “three stage” folding, is observed within the last two helices in bacteriorhodopsin (BR), a system that serves as a model membrane protein. We employ a reductionist approach to understand interplay of molecular factors underlying the apparent defiance. Leveraging available solution NMR structures, we construct, sample in silico, and analyze partially (PIn) and fully inserted (FIn) BR membrane states. The membrane lateral C-terminal helix (CH) in PIn is markedly prone to transient structural distortions over microsecond timescales; a disorder prone region (DPR) is thereby identified. While clear transmembrane propensities are not acquired, the distortions induce alterations in local membrane curvature and area per lipid. Importantly, energetic decompositions reveal that overall, the N-terminal helix (NH) is thermodynamically more stable in the PIn. Higher overall stability of the FIn arises from favorable interactions between the NH and the CH. Our results establish lack of spontaneous transition of the PIn to the FIn, and attributes their partitioning to barriers that exceed those accessible with thermal fluctuations. This work paves the way for further detailed studies aimed at determining the thermo-kinetic roles of the initial five helices, or complementary external factors, in complete helical folding and insertion in BR. We comment that complementing such efforts with the growing field of machine learning assisted energy landscape searches may offer unprecedented insights.
期刊介绍:
BBA Biomembranes has its main focus on membrane structure, function and biomolecular organization, membrane proteins, receptors, channels and anchors, fluidity and composition, model membranes and liposomes, membrane surface studies and ligand interactions, transport studies, and membrane dynamics.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
