Jerry Gao, Maxwell Tong, Chinkyu Lee, Jacek Gaertig, Thibault Legal, Khanh Huy Bui
{"title":"DomainFit:利用 AlphaFold2 预测模型识别中分辨率低温电子显微镜图中的蛋白质结构域","authors":"Jerry Gao, Maxwell Tong, Chinkyu Lee, Jacek Gaertig, Thibault Legal, Khanh Huy Bui","doi":"10.1016/j.str.2024.04.017","DOIUrl":null,"url":null,"abstract":"<p>Cryoelectron microscopy (cryo-EM) has revolutionized the structural determination of macromolecular complexes. With the paradigm shift to structure determination of highly complex endogenous macromolecular complexes <em>ex vivo</em> and <em>in situ</em> structural biology, there are an increasing number of structures of native complexes. These complexes often contain unidentified proteins, related to different cellular states or processes. Identifying proteins at resolutions lower than 4 Å remains challenging because side chains cannot be visualized reliably. Here, we present DomainFit, a program for semi-automated domain-level protein identification from cryo-EM maps, particularly at resolutions lower than 4 Å. By fitting domains from AlphaFold2-predicted models into cryo-EM maps, the program performs statistical analyses and attempts to identify the domains and protein candidates forming the density. Using DomainFit, we identified two microtubule inner proteins, one of which contains a CCDC81 domain and is exclusively localized in the proximal region of the doublet microtubule in <em>Tetrahymena thermophila</em>.</p>","PeriodicalId":22168,"journal":{"name":"Structure","volume":"9 1","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2024-05-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DomainFit: Identification of protein domains in cryo-EM maps at intermediate resolution using AlphaFold2-predicted models\",\"authors\":\"Jerry Gao, Maxwell Tong, Chinkyu Lee, Jacek Gaertig, Thibault Legal, Khanh Huy Bui\",\"doi\":\"10.1016/j.str.2024.04.017\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Cryoelectron microscopy (cryo-EM) has revolutionized the structural determination of macromolecular complexes. With the paradigm shift to structure determination of highly complex endogenous macromolecular complexes <em>ex vivo</em> and <em>in situ</em> structural biology, there are an increasing number of structures of native complexes. These complexes often contain unidentified proteins, related to different cellular states or processes. Identifying proteins at resolutions lower than 4 Å remains challenging because side chains cannot be visualized reliably. Here, we present DomainFit, a program for semi-automated domain-level protein identification from cryo-EM maps, particularly at resolutions lower than 4 Å. By fitting domains from AlphaFold2-predicted models into cryo-EM maps, the program performs statistical analyses and attempts to identify the domains and protein candidates forming the density. Using DomainFit, we identified two microtubule inner proteins, one of which contains a CCDC81 domain and is exclusively localized in the proximal region of the doublet microtubule in <em>Tetrahymena thermophila</em>.</p>\",\"PeriodicalId\":22168,\"journal\":{\"name\":\"Structure\",\"volume\":\"9 1\",\"pages\":\"\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-05-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Structure\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.str.2024.04.017\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structure","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.str.2024.04.017","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
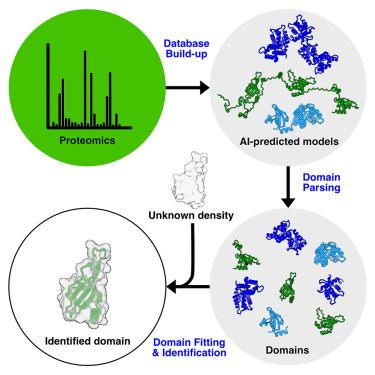
冷冻电镜(cryo-EM)为大分子复合物的结构测定带来了革命性的变化。随着体内外高难度内源大分子复合物结构测定和原位结构生物学模式的转变,原生复合物的结构越来越多。这些复合物通常含有与不同细胞状态或过程相关的未识别蛋白质。由于侧链不能被可靠地可视化,因此以低于 4 Å 的分辨率识别蛋白质仍然具有挑战性。通过将 AlphaFold2 预测模型中的结构域拟合到低温电子显微镜图中,该程序会执行统计分析,并尝试识别形成密度的结构域和候选蛋白质。利用 DomainFit,我们确定了两种微管内部蛋白,其中一种含有 CCDC81 结构域,专门定位于嗜热四膜虫双微管的近端区域。
DomainFit: Identification of protein domains in cryo-EM maps at intermediate resolution using AlphaFold2-predicted models
Cryoelectron microscopy (cryo-EM) has revolutionized the structural determination of macromolecular complexes. With the paradigm shift to structure determination of highly complex endogenous macromolecular complexes ex vivo and in situ structural biology, there are an increasing number of structures of native complexes. These complexes often contain unidentified proteins, related to different cellular states or processes. Identifying proteins at resolutions lower than 4 Å remains challenging because side chains cannot be visualized reliably. Here, we present DomainFit, a program for semi-automated domain-level protein identification from cryo-EM maps, particularly at resolutions lower than 4 Å. By fitting domains from AlphaFold2-predicted models into cryo-EM maps, the program performs statistical analyses and attempts to identify the domains and protein candidates forming the density. Using DomainFit, we identified two microtubule inner proteins, one of which contains a CCDC81 domain and is exclusively localized in the proximal region of the doublet microtubule in Tetrahymena thermophila.
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
