Yuanqi Du, Arian R. Jamasb, Jeff Guo, Tianfan Fu, Charles Harris, Yingheng Wang, Chenru Duan, Pietro Liò, Philippe Schwaller, Tom L. Blundell
{"title":"机器学习辅助生成分子设计","authors":"Yuanqi Du, Arian R. Jamasb, Jeff Guo, Tianfan Fu, Charles Harris, Yingheng Wang, Chenru Duan, Pietro Liò, Philippe Schwaller, Tom L. Blundell","doi":"10.1038/s42256-024-00843-5","DOIUrl":null,"url":null,"abstract":"Machine learning has provided a means to accelerate early-stage drug discovery by combining molecule generation and filtering steps in a single architecture that leverages the experience and design preferences of medicinal chemists. However, designing machine learning models that can achieve this on the fly to the satisfaction of medicinal chemists remains a challenge owing to the enormous search space. Researchers have addressed de novo design of molecules by decomposing the problem into a series of tasks determined by design criteria. Here we provide a comprehensive overview of the current state of the art in molecular design using machine learning models as well as important design decisions, such as the choice of molecular representations, generative methods and optimization strategies. Subsequently, we present a collection of practical applications in which the reviewed methodologies have been experimentally validated, encompassing both academic and industrial efforts. Finally, we draw attention to the theoretical, computational and empirical challenges in deploying generative machine learning and highlight future opportunities to better align such approaches to achieve realistic drug discovery end points. Data-driven generative methods have the potential to greatly facilitate molecular design tasks for drug design.","PeriodicalId":48533,"journal":{"name":"Nature Machine Intelligence","volume":"6 6","pages":"589-604"},"PeriodicalIF":29.8000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine learning-aided generative molecular design\",\"authors\":\"Yuanqi Du, Arian R. Jamasb, Jeff Guo, Tianfan Fu, Charles Harris, Yingheng Wang, Chenru Duan, Pietro Liò, Philippe Schwaller, Tom L. Blundell\",\"doi\":\"10.1038/s42256-024-00843-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Machine learning has provided a means to accelerate early-stage drug discovery by combining molecule generation and filtering steps in a single architecture that leverages the experience and design preferences of medicinal chemists. However, designing machine learning models that can achieve this on the fly to the satisfaction of medicinal chemists remains a challenge owing to the enormous search space. Researchers have addressed de novo design of molecules by decomposing the problem into a series of tasks determined by design criteria. Here we provide a comprehensive overview of the current state of the art in molecular design using machine learning models as well as important design decisions, such as the choice of molecular representations, generative methods and optimization strategies. Subsequently, we present a collection of practical applications in which the reviewed methodologies have been experimentally validated, encompassing both academic and industrial efforts. Finally, we draw attention to the theoretical, computational and empirical challenges in deploying generative machine learning and highlight future opportunities to better align such approaches to achieve realistic drug discovery end points. Data-driven generative methods have the potential to greatly facilitate molecular design tasks for drug design.\",\"PeriodicalId\":48533,\"journal\":{\"name\":\"Nature Machine Intelligence\",\"volume\":\"6 6\",\"pages\":\"589-604\"},\"PeriodicalIF\":29.8000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Machine Intelligence\",\"FirstCategoryId\":\"94\",\"ListUrlMain\":\"https://www.nature.com/articles/s42256-024-00843-5\",\"RegionNum\":1,\"RegionCategory\":\"计算机科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Machine Intelligence","FirstCategoryId":"94","ListUrlMain":"https://www.nature.com/articles/s42256-024-00843-5","RegionNum":1,"RegionCategory":"计算机科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE","Score":null,"Total":0}
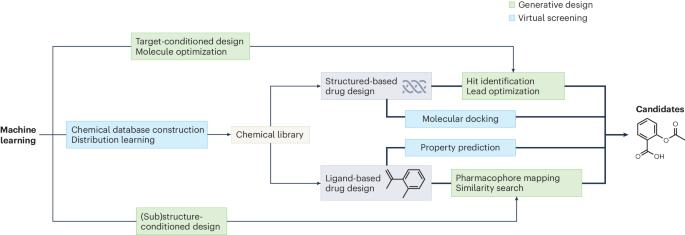
Machine learning has provided a means to accelerate early-stage drug discovery by combining molecule generation and filtering steps in a single architecture that leverages the experience and design preferences of medicinal chemists. However, designing machine learning models that can achieve this on the fly to the satisfaction of medicinal chemists remains a challenge owing to the enormous search space. Researchers have addressed de novo design of molecules by decomposing the problem into a series of tasks determined by design criteria. Here we provide a comprehensive overview of the current state of the art in molecular design using machine learning models as well as important design decisions, such as the choice of molecular representations, generative methods and optimization strategies. Subsequently, we present a collection of practical applications in which the reviewed methodologies have been experimentally validated, encompassing both academic and industrial efforts. Finally, we draw attention to the theoretical, computational and empirical challenges in deploying generative machine learning and highlight future opportunities to better align such approaches to achieve realistic drug discovery end points. Data-driven generative methods have the potential to greatly facilitate molecular design tasks for drug design.
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
