Maria Boulougouri, Pierre Vandergheynst, Daniel Probst
{"title":"分子集表示学习","authors":"Maria Boulougouri, Pierre Vandergheynst, Daniel Probst","doi":"10.1038/s42256-024-00856-0","DOIUrl":null,"url":null,"abstract":"Computational representation of molecules can take many forms, including graphs, string encodings of graphs, binary vectors or learned embeddings in the form of real-valued vectors. These representations are then used in downstream classification and regression tasks using a wide range of machine learning models. However, existing models come with limitations, such as the requirement for clearly defined chemical bonds, which often do not represent the true underlying nature of a molecule. Here we propose a framework for molecular machine learning tasks based on set representation learning. We show that learning on sets of atom invariants alone reaches the performance of state-of-the-art graph-based models on the most-used chemical benchmark datasets and that introducing a set representation layer into graph neural networks can surpass the performance of established methods in the domains of chemistry, biology and material science. We introduce specialized set representation-based neural network architectures for reaction-yield and protein–ligand binding-affinity prediction. Overall, we show that the technique we denote molecular set representation learning is both an alternative and an extension to graph neural network architectures for machine learning tasks on molecules, molecule complexes and chemical reactions. Machine learning methods for molecule predictions use various representations of molecules such as in the form of strings or graphs. As an extension of graph representation learning, Probst and colleagues propose to represent a molecule as a set of atoms, to better capture the underlying chemical nature, and demonstrate improved performance in a range of machine learning tasks.","PeriodicalId":48533,"journal":{"name":"Nature Machine Intelligence","volume":"6 7","pages":"754-763"},"PeriodicalIF":23.9000,"publicationDate":"2024-07-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s42256-024-00856-0.pdf","citationCount":"0","resultStr":"{\"title\":\"Molecular set representation learning\",\"authors\":\"Maria Boulougouri, Pierre Vandergheynst, Daniel Probst\",\"doi\":\"10.1038/s42256-024-00856-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Computational representation of molecules can take many forms, including graphs, string encodings of graphs, binary vectors or learned embeddings in the form of real-valued vectors. These representations are then used in downstream classification and regression tasks using a wide range of machine learning models. However, existing models come with limitations, such as the requirement for clearly defined chemical bonds, which often do not represent the true underlying nature of a molecule. Here we propose a framework for molecular machine learning tasks based on set representation learning. We show that learning on sets of atom invariants alone reaches the performance of state-of-the-art graph-based models on the most-used chemical benchmark datasets and that introducing a set representation layer into graph neural networks can surpass the performance of established methods in the domains of chemistry, biology and material science. We introduce specialized set representation-based neural network architectures for reaction-yield and protein–ligand binding-affinity prediction. Overall, we show that the technique we denote molecular set representation learning is both an alternative and an extension to graph neural network architectures for machine learning tasks on molecules, molecule complexes and chemical reactions. Machine learning methods for molecule predictions use various representations of molecules such as in the form of strings or graphs. As an extension of graph representation learning, Probst and colleagues propose to represent a molecule as a set of atoms, to better capture the underlying chemical nature, and demonstrate improved performance in a range of machine learning tasks.\",\"PeriodicalId\":48533,\"journal\":{\"name\":\"Nature Machine Intelligence\",\"volume\":\"6 7\",\"pages\":\"754-763\"},\"PeriodicalIF\":23.9000,\"publicationDate\":\"2024-07-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s42256-024-00856-0.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Machine Intelligence\",\"FirstCategoryId\":\"94\",\"ListUrlMain\":\"https://www.nature.com/articles/s42256-024-00856-0\",\"RegionNum\":1,\"RegionCategory\":\"计算机科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Machine Intelligence","FirstCategoryId":"94","ListUrlMain":"https://www.nature.com/articles/s42256-024-00856-0","RegionNum":1,"RegionCategory":"计算机科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE","Score":null,"Total":0}
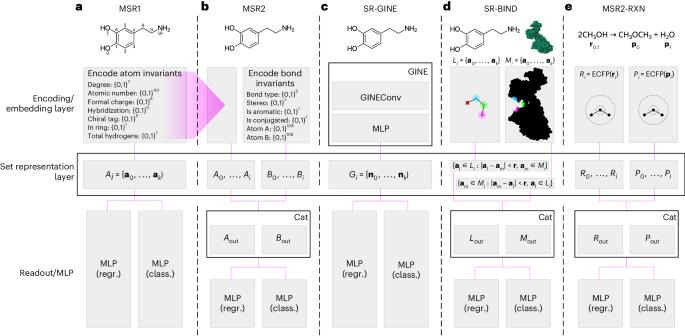
Computational representation of molecules can take many forms, including graphs, string encodings of graphs, binary vectors or learned embeddings in the form of real-valued vectors. These representations are then used in downstream classification and regression tasks using a wide range of machine learning models. However, existing models come with limitations, such as the requirement for clearly defined chemical bonds, which often do not represent the true underlying nature of a molecule. Here we propose a framework for molecular machine learning tasks based on set representation learning. We show that learning on sets of atom invariants alone reaches the performance of state-of-the-art graph-based models on the most-used chemical benchmark datasets and that introducing a set representation layer into graph neural networks can surpass the performance of established methods in the domains of chemistry, biology and material science. We introduce specialized set representation-based neural network architectures for reaction-yield and protein–ligand binding-affinity prediction. Overall, we show that the technique we denote molecular set representation learning is both an alternative and an extension to graph neural network architectures for machine learning tasks on molecules, molecule complexes and chemical reactions. Machine learning methods for molecule predictions use various representations of molecules such as in the form of strings or graphs. As an extension of graph representation learning, Probst and colleagues propose to represent a molecule as a set of atoms, to better capture the underlying chemical nature, and demonstrate improved performance in a range of machine learning tasks.
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
