Emily R Hannon, Carmen J Marsit, Arlene E Dent, Paula Embury, Sidney Ogolla, David Midem, Scott M Williams, James W Kazura
{"title":"转录组和基于 DNA 甲基化的细胞类型解旋对差异基因表达和差异甲基化的估计结果相似。","authors":"Emily R Hannon, Carmen J Marsit, Arlene E Dent, Paula Embury, Sidney Ogolla, David Midem, Scott M Williams, James W Kazura","doi":"10.1186/s13040-024-00374-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Changing cell-type proportions can confound studies of differential gene expression or DNA methylation (DNAm) from peripheral blood mononuclear cells (PBMCs). We examined how cell-type proportions derived from the transcriptome versus the methylome (DNAm) influence estimates of differentially expressed genes (DEGs) and differentially methylated positions (DMPs).</p><p><strong>Methods: </strong>Transcriptome and DNAm data were obtained from PBMC RNA and DNA of Kenyan children (n = 8) before, during, and 6 weeks following uncomplicated malaria. DEGs and DMPs between time points were detected using cell-type adjusted modeling with Cibersortx or IDOL, respectively.</p><p><strong>Results: </strong>Most major cell types and principal components had moderate to high correlation between the two deconvolution methods (r = 0.60-0.96). Estimates of cell-type proportions and DEGs or DMPs were largely unaffected by the method, with the greatest discrepancy in the estimation of neutrophils.</p><p><strong>Conclusion: </strong>Variation in cell-type proportions is captured similarly by both transcriptomic and methylome deconvolution methods for most major cell types.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"17 1","pages":"21"},"PeriodicalIF":6.1000,"publicationDate":"2024-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11241886/pdf/","citationCount":"0","resultStr":"{\"title\":\"Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation.\",\"authors\":\"Emily R Hannon, Carmen J Marsit, Arlene E Dent, Paula Embury, Sidney Ogolla, David Midem, Scott M Williams, James W Kazura\",\"doi\":\"10.1186/s13040-024-00374-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Changing cell-type proportions can confound studies of differential gene expression or DNA methylation (DNAm) from peripheral blood mononuclear cells (PBMCs). We examined how cell-type proportions derived from the transcriptome versus the methylome (DNAm) influence estimates of differentially expressed genes (DEGs) and differentially methylated positions (DMPs).</p><p><strong>Methods: </strong>Transcriptome and DNAm data were obtained from PBMC RNA and DNA of Kenyan children (n = 8) before, during, and 6 weeks following uncomplicated malaria. DEGs and DMPs between time points were detected using cell-type adjusted modeling with Cibersortx or IDOL, respectively.</p><p><strong>Results: </strong>Most major cell types and principal components had moderate to high correlation between the two deconvolution methods (r = 0.60-0.96). Estimates of cell-type proportions and DEGs or DMPs were largely unaffected by the method, with the greatest discrepancy in the estimation of neutrophils.</p><p><strong>Conclusion: </strong>Variation in cell-type proportions is captured similarly by both transcriptomic and methylome deconvolution methods for most major cell types.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\"17 1\",\"pages\":\"21\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2024-07-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11241886/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-024-00374-0\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-024-00374-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation.
Background: Changing cell-type proportions can confound studies of differential gene expression or DNA methylation (DNAm) from peripheral blood mononuclear cells (PBMCs). We examined how cell-type proportions derived from the transcriptome versus the methylome (DNAm) influence estimates of differentially expressed genes (DEGs) and differentially methylated positions (DMPs).
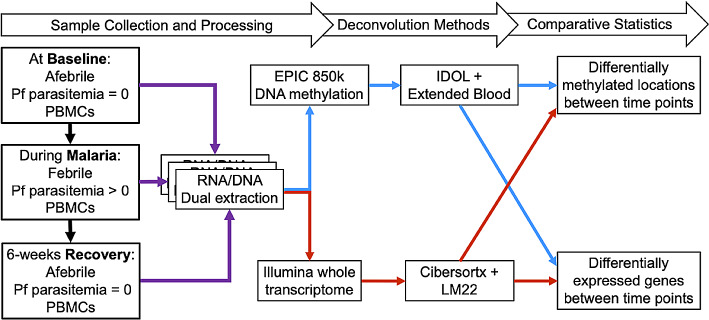
Methods: Transcriptome and DNAm data were obtained from PBMC RNA and DNA of Kenyan children (n = 8) before, during, and 6 weeks following uncomplicated malaria. DEGs and DMPs between time points were detected using cell-type adjusted modeling with Cibersortx or IDOL, respectively.
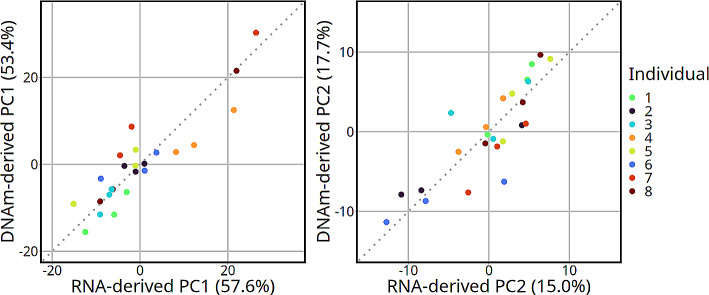
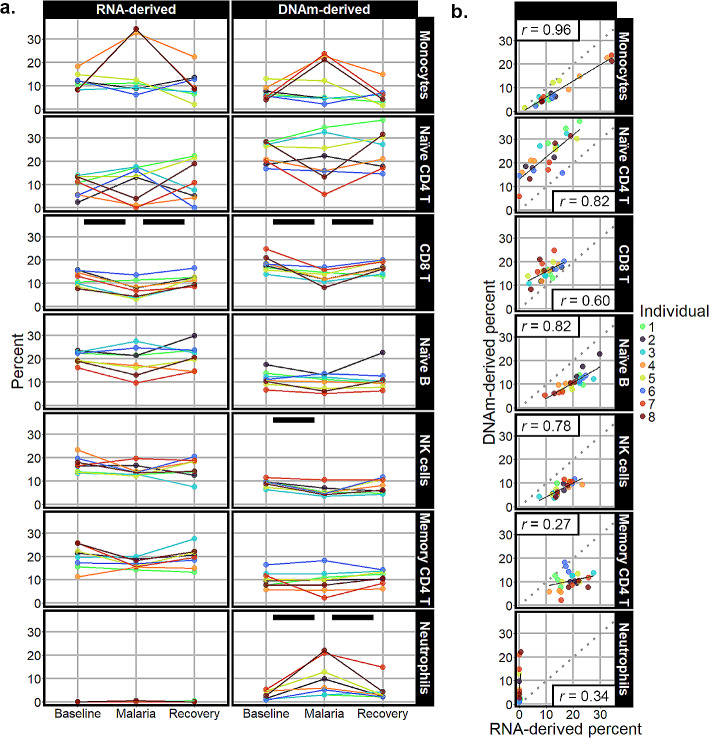
Results: Most major cell types and principal components had moderate to high correlation between the two deconvolution methods (r = 0.60-0.96). Estimates of cell-type proportions and DEGs or DMPs were largely unaffected by the method, with the greatest discrepancy in the estimation of neutrophils.
Conclusion: Variation in cell-type proportions is captured similarly by both transcriptomic and methylome deconvolution methods for most major cell types.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
