Rebaz Anwar Omer, Yousif Hussein Azeez, Rebaz Obaid Kareem, Lana Omer Ahmed, Damir A. Safin
{"title":"香豆素衍生物潜在缓蚀特性的 DFT 和蒙特卡罗模拟联合研究。","authors":"Rebaz Anwar Omer, Yousif Hussein Azeez, Rebaz Obaid Kareem, Lana Omer Ahmed, Damir A. Safin","doi":"10.1007/s00894-024-06090-0","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Corrosion, the degradation of materials due to chemical reactions with their environment presents significant challenges both economically and environmentally. It affects various industries, including construction, transportation, and manufacturing, leading to equipment failures, safety hazards, and increased maintenance costs. Coumarin derivatives have shown promise due to their inherent chemical properties and potential for biodegradability. In this study, a series of the coumarin derivatives were examined in silico to reveal their potential corrosion inhibition properties toward the Fe(110) and Cu(111) surfaces. The compounds investigated include coumarin (2<i>H</i>-chromen-2-one, <b>1</b>), furanocoumarin (7<i>H</i>-furo[3,2-<i>g</i>]chromen-7-one, <b>2</b>), dihydrofurano coumarin (2,3-dihydro-7<i>H</i>-furo[3,2-<i>g</i>]chromen-7-one, <b>3</b>), pyrano coumarin–linear type (8,8-dimethyl-2<i>H</i>,8<i>H</i>-pyrano[3,2-<i>g</i>]chromen-2-one, <b>4</b>), pyrano coumarin–angular type (8,8-dimethyl-2<i>H</i>,8<i>H</i>-pyrano[2,3-<i>f</i>]chromen-2-one, <b>5</b>), bicoumarin (3,3'-methylenebis(2<i>H</i>-chromen-2-one), <b>6</b>), and phenyl coumarin (4-phenyl-2<i>H</i>-chromen-2-one, <b>7</b>). The findings suggest that the bicoumarin derivative <b>6</b> exhibits the lowest adsorption energy with the Fe(110) surface, while the same energy absolute value is about two times lower for the Cu(111) surface. This is due to the formation of a planar configuration of a molecule of <b>6</b> on the metal surfaces with the participation of both coumarin fragments upon interacting with the Fe(110) surface, while one coumarin fragment interacts with the Cu(111) surface.</p><h3>Methods</h3><p>Density functional theory (DFT) calculations were employed to study the electronic properties of the coumarin derivatives. The specific computational method used was B3LYP, a hybrid functional that combines with the 6–311 + + G(d,p) basis set. Each coumarin derivative was first subjected to a geometry optimization to find the most stable molecular structure. Electronic properties, dipole moments, and molecular electrostatic potential surfaces were calculated. The Monte Carlo simulations were used to model the adsorption behavior of the coumarin derivatives on metal surfaces, namely, Fe(110) and Cu(111). These simulations allowed to visualize interaction of the studied molecules with the metal surfaces, which is crucial for their function as corrosion inhibitors. The present study provides a comprehensive understanding of the corrosion inhibition potential of the applied coumarin derivatives. The insights gained from these methods can inform the development of effective, sustainable corrosion inhibitors that are both environmentally friendly and highly efficient.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Combined DFT and Monte Carlo simulation studies of potential corrosion inhibition properties of coumarin derivatives\",\"authors\":\"Rebaz Anwar Omer, Yousif Hussein Azeez, Rebaz Obaid Kareem, Lana Omer Ahmed, Damir A. Safin\",\"doi\":\"10.1007/s00894-024-06090-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Corrosion, the degradation of materials due to chemical reactions with their environment presents significant challenges both economically and environmentally. It affects various industries, including construction, transportation, and manufacturing, leading to equipment failures, safety hazards, and increased maintenance costs. Coumarin derivatives have shown promise due to their inherent chemical properties and potential for biodegradability. In this study, a series of the coumarin derivatives were examined in silico to reveal their potential corrosion inhibition properties toward the Fe(110) and Cu(111) surfaces. The compounds investigated include coumarin (2<i>H</i>-chromen-2-one, <b>1</b>), furanocoumarin (7<i>H</i>-furo[3,2-<i>g</i>]chromen-7-one, <b>2</b>), dihydrofurano coumarin (2,3-dihydro-7<i>H</i>-furo[3,2-<i>g</i>]chromen-7-one, <b>3</b>), pyrano coumarin–linear type (8,8-dimethyl-2<i>H</i>,8<i>H</i>-pyrano[3,2-<i>g</i>]chromen-2-one, <b>4</b>), pyrano coumarin–angular type (8,8-dimethyl-2<i>H</i>,8<i>H</i>-pyrano[2,3-<i>f</i>]chromen-2-one, <b>5</b>), bicoumarin (3,3'-methylenebis(2<i>H</i>-chromen-2-one), <b>6</b>), and phenyl coumarin (4-phenyl-2<i>H</i>-chromen-2-one, <b>7</b>). The findings suggest that the bicoumarin derivative <b>6</b> exhibits the lowest adsorption energy with the Fe(110) surface, while the same energy absolute value is about two times lower for the Cu(111) surface. This is due to the formation of a planar configuration of a molecule of <b>6</b> on the metal surfaces with the participation of both coumarin fragments upon interacting with the Fe(110) surface, while one coumarin fragment interacts with the Cu(111) surface.</p><h3>Methods</h3><p>Density functional theory (DFT) calculations were employed to study the electronic properties of the coumarin derivatives. The specific computational method used was B3LYP, a hybrid functional that combines with the 6–311 + + G(d,p) basis set. Each coumarin derivative was first subjected to a geometry optimization to find the most stable molecular structure. Electronic properties, dipole moments, and molecular electrostatic potential surfaces were calculated. The Monte Carlo simulations were used to model the adsorption behavior of the coumarin derivatives on metal surfaces, namely, Fe(110) and Cu(111). These simulations allowed to visualize interaction of the studied molecules with the metal surfaces, which is crucial for their function as corrosion inhibitors. The present study provides a comprehensive understanding of the corrosion inhibition potential of the applied coumarin derivatives. The insights gained from these methods can inform the development of effective, sustainable corrosion inhibitors that are both environmentally friendly and highly efficient.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 8\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-07-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06090-0\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06090-0","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Combined DFT and Monte Carlo simulation studies of potential corrosion inhibition properties of coumarin derivatives
Context
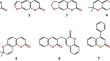
Corrosion, the degradation of materials due to chemical reactions with their environment presents significant challenges both economically and environmentally. It affects various industries, including construction, transportation, and manufacturing, leading to equipment failures, safety hazards, and increased maintenance costs. Coumarin derivatives have shown promise due to their inherent chemical properties and potential for biodegradability. In this study, a series of the coumarin derivatives were examined in silico to reveal their potential corrosion inhibition properties toward the Fe(110) and Cu(111) surfaces. The compounds investigated include coumarin (2H-chromen-2-one, 1), furanocoumarin (7H-furo[3,2-g]chromen-7-one, 2), dihydrofurano coumarin (2,3-dihydro-7H-furo[3,2-g]chromen-7-one, 3), pyrano coumarin–linear type (8,8-dimethyl-2H,8H-pyrano[3,2-g]chromen-2-one, 4), pyrano coumarin–angular type (8,8-dimethyl-2H,8H-pyrano[2,3-f]chromen-2-one, 5), bicoumarin (3,3'-methylenebis(2H-chromen-2-one), 6), and phenyl coumarin (4-phenyl-2H-chromen-2-one, 7). The findings suggest that the bicoumarin derivative 6 exhibits the lowest adsorption energy with the Fe(110) surface, while the same energy absolute value is about two times lower for the Cu(111) surface. This is due to the formation of a planar configuration of a molecule of 6 on the metal surfaces with the participation of both coumarin fragments upon interacting with the Fe(110) surface, while one coumarin fragment interacts with the Cu(111) surface.
Methods
Density functional theory (DFT) calculations were employed to study the electronic properties of the coumarin derivatives. The specific computational method used was B3LYP, a hybrid functional that combines with the 6–311 + + G(d,p) basis set. Each coumarin derivative was first subjected to a geometry optimization to find the most stable molecular structure. Electronic properties, dipole moments, and molecular electrostatic potential surfaces were calculated. The Monte Carlo simulations were used to model the adsorption behavior of the coumarin derivatives on metal surfaces, namely, Fe(110) and Cu(111). These simulations allowed to visualize interaction of the studied molecules with the metal surfaces, which is crucial for their function as corrosion inhibitors. The present study provides a comprehensive understanding of the corrosion inhibition potential of the applied coumarin derivatives. The insights gained from these methods can inform the development of effective, sustainable corrosion inhibitors that are both environmentally friendly and highly efficient.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
