Victoria Defilippi, Juli Petereit, Valerie J. L. Handlos, Lucia Notterpek
{"title":"定量蛋白质组学揭示了神经病理性神经中已知和以前未认识到的变化。","authors":"Victoria Defilippi, Juli Petereit, Valerie J. L. Handlos, Lucia Notterpek","doi":"10.1111/jnc.16189","DOIUrl":null,"url":null,"abstract":"<p>Charcot–Marie–Tooth disease type 1E (CMT1E) is an inherited autosomal dominant peripheral neuropathy caused by mutations in the <i>peripheral myelin protein 22</i> (<i>PMP22</i>) gene. The identical leucine-to-proline (L16P) amino acid substitution in PMP22 is carried by the Trembler J (TrJ) mouse and is found in CMT1E patients presenting with early-onset disease. Peripheral nerves of patients diagnosed with CMT1E display a complex and varied histopathology, including Schwann cell hyperproliferation, abnormally thin myelin, axonal degeneration, and subaxonal morphological changes. Here, we have taken an unbiased data-independent analysis (DIA) mass spectrometry (MS) approach to quantify proteins from nerves of 3-week-old, age and genetic strain-matched wild-type (Wt) and heterozygous TrJ mice. Nerve proteins were dissolved in lysis buffer and digested into peptide fragments, and protein groups were quantified by liquid chromatography-mass spectrometry (LC–MS). A linear model determined statistically significant differences between the study groups, and proteins with an adjusted p-value of less than 0.05 were deemed significant. This untargeted proteomics approach identified 3759 quality-controlled protein groups, of which 884 demonstrated differential expression between the two genotypes. Gene ontology (GO) terms related to myelin and myelin maintenance confirm published data while revealing a previously undetected prominent decrease in peripheral myelin protein 2. The dataset corroborates the described pathophysiology of TrJ nerves, including elevated activity in the proteasome-lysosomal pathways, alterations in protein trafficking, and an increase in three macrophage-associated proteins. Previously unrecognized perturbations in RNA processing pathways and GO terms were also discovered. Proteomic abnormalities that overlap with other human neurological disorders besides CMT include Lafora Disease and Amyotrophic Lateral Sclerosis. Overall, this study confirms and extends current knowledge on the cellular pathophysiology in TrJ neuropathic nerves and provides novel insights for future examinations. Recognition of shared pathomechanisms across discrete neurological disorders offers opportunities for innovative disease-modifying therapeutics that could be effective for distinct neuropathies.\n <figure>\n <div><picture>\n <source></source></picture><p></p>\n </div>\n </figure></p>","PeriodicalId":16527,"journal":{"name":"Journal of Neurochemistry","volume":"168 9","pages":"3154-3170"},"PeriodicalIF":4.0000,"publicationDate":"2024-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jnc.16189","citationCount":"0","resultStr":"{\"title\":\"Quantitative proteomics unveils known and previously unrecognized alterations in neuropathic nerves\",\"authors\":\"Victoria Defilippi, Juli Petereit, Valerie J. L. Handlos, Lucia Notterpek\",\"doi\":\"10.1111/jnc.16189\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Charcot–Marie–Tooth disease type 1E (CMT1E) is an inherited autosomal dominant peripheral neuropathy caused by mutations in the <i>peripheral myelin protein 22</i> (<i>PMP22</i>) gene. The identical leucine-to-proline (L16P) amino acid substitution in PMP22 is carried by the Trembler J (TrJ) mouse and is found in CMT1E patients presenting with early-onset disease. Peripheral nerves of patients diagnosed with CMT1E display a complex and varied histopathology, including Schwann cell hyperproliferation, abnormally thin myelin, axonal degeneration, and subaxonal morphological changes. Here, we have taken an unbiased data-independent analysis (DIA) mass spectrometry (MS) approach to quantify proteins from nerves of 3-week-old, age and genetic strain-matched wild-type (Wt) and heterozygous TrJ mice. Nerve proteins were dissolved in lysis buffer and digested into peptide fragments, and protein groups were quantified by liquid chromatography-mass spectrometry (LC–MS). A linear model determined statistically significant differences between the study groups, and proteins with an adjusted p-value of less than 0.05 were deemed significant. This untargeted proteomics approach identified 3759 quality-controlled protein groups, of which 884 demonstrated differential expression between the two genotypes. Gene ontology (GO) terms related to myelin and myelin maintenance confirm published data while revealing a previously undetected prominent decrease in peripheral myelin protein 2. The dataset corroborates the described pathophysiology of TrJ nerves, including elevated activity in the proteasome-lysosomal pathways, alterations in protein trafficking, and an increase in three macrophage-associated proteins. Previously unrecognized perturbations in RNA processing pathways and GO terms were also discovered. Proteomic abnormalities that overlap with other human neurological disorders besides CMT include Lafora Disease and Amyotrophic Lateral Sclerosis. Overall, this study confirms and extends current knowledge on the cellular pathophysiology in TrJ neuropathic nerves and provides novel insights for future examinations. Recognition of shared pathomechanisms across discrete neurological disorders offers opportunities for innovative disease-modifying therapeutics that could be effective for distinct neuropathies.\\n <figure>\\n <div><picture>\\n <source></source></picture><p></p>\\n </div>\\n </figure></p>\",\"PeriodicalId\":16527,\"journal\":{\"name\":\"Journal of Neurochemistry\",\"volume\":\"168 9\",\"pages\":\"3154-3170\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2024-07-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jnc.16189\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neurochemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/jnc.16189\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurochemistry","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jnc.16189","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Quantitative proteomics unveils known and previously unrecognized alterations in neuropathic nerves
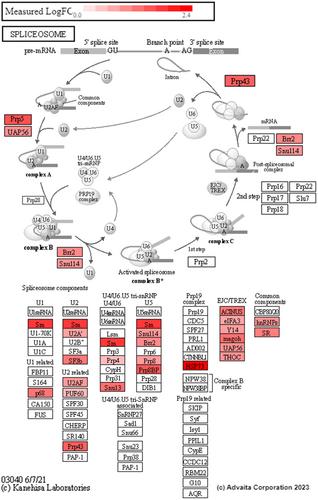
Charcot–Marie–Tooth disease type 1E (CMT1E) is an inherited autosomal dominant peripheral neuropathy caused by mutations in the peripheral myelin protein 22 (PMP22) gene. The identical leucine-to-proline (L16P) amino acid substitution in PMP22 is carried by the Trembler J (TrJ) mouse and is found in CMT1E patients presenting with early-onset disease. Peripheral nerves of patients diagnosed with CMT1E display a complex and varied histopathology, including Schwann cell hyperproliferation, abnormally thin myelin, axonal degeneration, and subaxonal morphological changes. Here, we have taken an unbiased data-independent analysis (DIA) mass spectrometry (MS) approach to quantify proteins from nerves of 3-week-old, age and genetic strain-matched wild-type (Wt) and heterozygous TrJ mice. Nerve proteins were dissolved in lysis buffer and digested into peptide fragments, and protein groups were quantified by liquid chromatography-mass spectrometry (LC–MS). A linear model determined statistically significant differences between the study groups, and proteins with an adjusted p-value of less than 0.05 were deemed significant. This untargeted proteomics approach identified 3759 quality-controlled protein groups, of which 884 demonstrated differential expression between the two genotypes. Gene ontology (GO) terms related to myelin and myelin maintenance confirm published data while revealing a previously undetected prominent decrease in peripheral myelin protein 2. The dataset corroborates the described pathophysiology of TrJ nerves, including elevated activity in the proteasome-lysosomal pathways, alterations in protein trafficking, and an increase in three macrophage-associated proteins. Previously unrecognized perturbations in RNA processing pathways and GO terms were also discovered. Proteomic abnormalities that overlap with other human neurological disorders besides CMT include Lafora Disease and Amyotrophic Lateral Sclerosis. Overall, this study confirms and extends current knowledge on the cellular pathophysiology in TrJ neuropathic nerves and provides novel insights for future examinations. Recognition of shared pathomechanisms across discrete neurological disorders offers opportunities for innovative disease-modifying therapeutics that could be effective for distinct neuropathies.
期刊介绍:
Journal of Neurochemistry focuses on molecular, cellular and biochemical aspects of the nervous system, the pathogenesis of neurological disorders and the development of disease specific biomarkers. It is devoted to the prompt publication of original findings of the highest scientific priority and value that provide novel mechanistic insights, represent a clear advance over previous studies and have the potential to generate exciting future research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
