Aly Elezaby, Amanda J. Lin, Vijith Vijayan, Suman Pokhrel, Benjamin R. Kraemer, Luiz R. G. Bechara, Isabel Larus, Junhui Sun, Valentina Baena, Zulfeqhar A. Syed, Elizabeth Murphy, Brian Glancy, Nicolai P. Ostberg, Bruno B. Queliconi, Juliane C. Campos, Julio C. B. Ferreira, Bereketeab Haileselassie, Daria Mochly-Rosen
{"title":"心肌肌钙蛋白 I 直接结合并抑制线粒体 ATP 合成酶,在缺血后心脏中发挥非典型作用","authors":"Aly Elezaby, Amanda J. Lin, Vijith Vijayan, Suman Pokhrel, Benjamin R. Kraemer, Luiz R. G. Bechara, Isabel Larus, Junhui Sun, Valentina Baena, Zulfeqhar A. Syed, Elizabeth Murphy, Brian Glancy, Nicolai P. Ostberg, Bruno B. Queliconi, Juliane C. Campos, Julio C. B. Ferreira, Bereketeab Haileselassie, Daria Mochly-Rosen","doi":"10.1038/s44161-024-00512-1","DOIUrl":null,"url":null,"abstract":"Cardiac troponin I (cTnI) is a key regulator of cardiomyocyte contraction. However, its role in mitochondria is unknown. Here we show that cTnI localized to mitochondria in the heart, inhibited mitochondrial functions when stably expressed in noncardiac cells and increased the opening of the mitochondrial permeability transition pore under oxidative stress. Direct, specific and saturable binding of cTnI to F1FO-ATP synthase was demonstrated in vitro using immune-captured ATP synthase and in cells using proximity ligation assay. cTnI binding doubled ATPase activity, whereas skeletal troponin I and several human pathogenic cTnI variants associated with familial hypertrophic cardiomyopathy did not. A rationally designed peptide, P888, inhibited cTnI binding to ATP synthase, inhibited cTnI-induced increase in ATPase activity in vitro and reduced cardiac injury following transient ischemia in vivo. We suggest that cTnI-bound ATP synthase results in lower ATP levels, and releasing this interaction during cardiac ischemia–reperfusion may increase the reservoir of functional mitochondria to reduce cardiac injury. Elezaby et al. show that cardiac troponin I interacts with mitochondrial ATP synthase to increase ATPase activity. Disrupting this interaction reduces cardiac damage following transient ischemia.","PeriodicalId":74245,"journal":{"name":"Nature cardiovascular research","volume":"3 8","pages":"987-1002"},"PeriodicalIF":12.6000,"publicationDate":"2024-07-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Cardiac troponin I directly binds and inhibits mitochondrial ATP synthase with a noncanonical role in the post-ischemic heart\",\"authors\":\"Aly Elezaby, Amanda J. Lin, Vijith Vijayan, Suman Pokhrel, Benjamin R. Kraemer, Luiz R. G. Bechara, Isabel Larus, Junhui Sun, Valentina Baena, Zulfeqhar A. Syed, Elizabeth Murphy, Brian Glancy, Nicolai P. Ostberg, Bruno B. Queliconi, Juliane C. Campos, Julio C. B. Ferreira, Bereketeab Haileselassie, Daria Mochly-Rosen\",\"doi\":\"10.1038/s44161-024-00512-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Cardiac troponin I (cTnI) is a key regulator of cardiomyocyte contraction. However, its role in mitochondria is unknown. Here we show that cTnI localized to mitochondria in the heart, inhibited mitochondrial functions when stably expressed in noncardiac cells and increased the opening of the mitochondrial permeability transition pore under oxidative stress. Direct, specific and saturable binding of cTnI to F1FO-ATP synthase was demonstrated in vitro using immune-captured ATP synthase and in cells using proximity ligation assay. cTnI binding doubled ATPase activity, whereas skeletal troponin I and several human pathogenic cTnI variants associated with familial hypertrophic cardiomyopathy did not. A rationally designed peptide, P888, inhibited cTnI binding to ATP synthase, inhibited cTnI-induced increase in ATPase activity in vitro and reduced cardiac injury following transient ischemia in vivo. We suggest that cTnI-bound ATP synthase results in lower ATP levels, and releasing this interaction during cardiac ischemia–reperfusion may increase the reservoir of functional mitochondria to reduce cardiac injury. Elezaby et al. show that cardiac troponin I interacts with mitochondrial ATP synthase to increase ATPase activity. Disrupting this interaction reduces cardiac damage following transient ischemia.\",\"PeriodicalId\":74245,\"journal\":{\"name\":\"Nature cardiovascular research\",\"volume\":\"3 8\",\"pages\":\"987-1002\"},\"PeriodicalIF\":12.6000,\"publicationDate\":\"2024-07-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature cardiovascular research\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s44161-024-00512-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CARDIAC & CARDIOVASCULAR SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature cardiovascular research","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s44161-024-00512-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
引用次数: 0
摘要
心肌肌钙蛋白 I(cTnI)是心肌细胞收缩的关键调节因子。然而,它在线粒体中的作用尚不清楚。在这里,我们发现 cTnI 定位于心脏线粒体,在非心脏细胞中稳定表达时会抑制线粒体功能,并在氧化应激下增加线粒体通透性转换孔的开放。cTnI 与 F1FO-ATP 合成酶的直接、特异和饱和结合在体外使用免疫捕获的 ATP 合成酶得到了证实,在细胞中使用近接试验也得到了证实。合理设计的多肽 P888 可抑制 cTnI 与 ATP 合成酶的结合,抑制体外 cTnI 诱导的 ATP 酶活性的增加,并减轻体内短暂缺血后的心脏损伤。我们认为,cTnI 与 ATP 合成酶结合会导致 ATP 水平降低,在心脏缺血再灌注过程中释放这种相互作用可能会增加功能线粒体库,从而减轻心脏损伤。Elezaby 等人的研究表明,心肌肌钙蛋白 I 与线粒体 ATP 合成酶相互作用,增加 ATP 酶的活性。破坏这种相互作用可减少短暂缺血后的心脏损伤。
Cardiac troponin I directly binds and inhibits mitochondrial ATP synthase with a noncanonical role in the post-ischemic heart
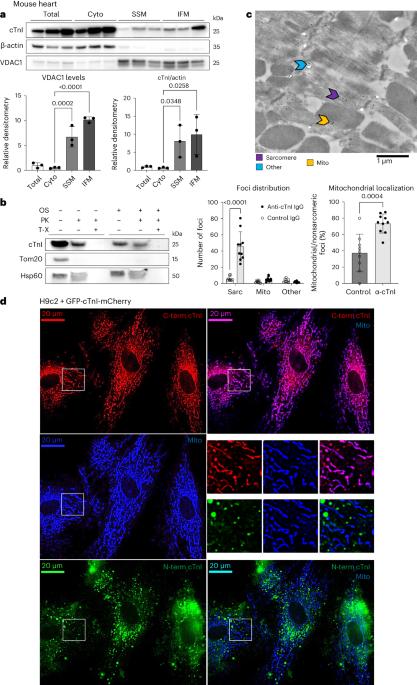
Cardiac troponin I (cTnI) is a key regulator of cardiomyocyte contraction. However, its role in mitochondria is unknown. Here we show that cTnI localized to mitochondria in the heart, inhibited mitochondrial functions when stably expressed in noncardiac cells and increased the opening of the mitochondrial permeability transition pore under oxidative stress. Direct, specific and saturable binding of cTnI to F1FO-ATP synthase was demonstrated in vitro using immune-captured ATP synthase and in cells using proximity ligation assay. cTnI binding doubled ATPase activity, whereas skeletal troponin I and several human pathogenic cTnI variants associated with familial hypertrophic cardiomyopathy did not. A rationally designed peptide, P888, inhibited cTnI binding to ATP synthase, inhibited cTnI-induced increase in ATPase activity in vitro and reduced cardiac injury following transient ischemia in vivo. We suggest that cTnI-bound ATP synthase results in lower ATP levels, and releasing this interaction during cardiac ischemia–reperfusion may increase the reservoir of functional mitochondria to reduce cardiac injury. Elezaby et al. show that cardiac troponin I interacts with mitochondrial ATP synthase to increase ATPase activity. Disrupting this interaction reduces cardiac damage following transient ischemia.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
