José Luis Cuevas Figueroa, Saravana Prakash Thirumuruganandham, Duncan John Mowbray, Alejandro Trejo Baños, Fernando Adán Serrano Orozco, Fabian Jimenez, Miguel Ojeda-Martínez
{"title":"富碳和富硅氢化圆碳化硅量子点中硅和碳替代缺陷及孔隙率的电子特性:一项 Ab-Initio 研究","authors":"José Luis Cuevas Figueroa, Saravana Prakash Thirumuruganandham, Duncan John Mowbray, Alejandro Trejo Baños, Fernando Adán Serrano Orozco, Fabian Jimenez, Miguel Ojeda-Martínez","doi":"10.1002/adts.202400637","DOIUrl":null,"url":null,"abstract":"<p>In this study, SiC quantum dots (SiC-QD's) are studied, and some roundish SiC-QD's with the incorporation of defects by removing a carbon or silicon atom are considered. Fourteen configurations are modeled in which the position of the silicon or carbon defect for each configuration is changed, considering that due to the chemical composition, it allows more Si atoms or more C atoms on the QD surface. All calculations are performed using the Density Functional Theory (DFT) methodology. The electronic exchange correlation is treated using the Generalized Gradient Approximation (GGA) with the Revised Perdew–Burke–Ernzerhof (RPBE) functional. The electronic energy levels of each configuration are calculated as well as the partial density of states to know the origin of the energy gap in each quantum dot. The final step is to analyze the energy formation to determine chemical stability.</p>","PeriodicalId":7219,"journal":{"name":"Advanced Theory and Simulations","volume":"7 11","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Electronic Properties of Si and C Substitutional Defects and Porosity in C-Rich and Si-Rich Hydrogenated Roundish SiC Quantum Dots: An Ab-Initio Study\",\"authors\":\"José Luis Cuevas Figueroa, Saravana Prakash Thirumuruganandham, Duncan John Mowbray, Alejandro Trejo Baños, Fernando Adán Serrano Orozco, Fabian Jimenez, Miguel Ojeda-Martínez\",\"doi\":\"10.1002/adts.202400637\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In this study, SiC quantum dots (SiC-QD's) are studied, and some roundish SiC-QD's with the incorporation of defects by removing a carbon or silicon atom are considered. Fourteen configurations are modeled in which the position of the silicon or carbon defect for each configuration is changed, considering that due to the chemical composition, it allows more Si atoms or more C atoms on the QD surface. All calculations are performed using the Density Functional Theory (DFT) methodology. The electronic exchange correlation is treated using the Generalized Gradient Approximation (GGA) with the Revised Perdew–Burke–Ernzerhof (RPBE) functional. The electronic energy levels of each configuration are calculated as well as the partial density of states to know the origin of the energy gap in each quantum dot. The final step is to analyze the energy formation to determine chemical stability.</p>\",\"PeriodicalId\":7219,\"journal\":{\"name\":\"Advanced Theory and Simulations\",\"volume\":\"7 11\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advanced Theory and Simulations\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://advanced.onlinelibrary.wiley.com/doi/10.1002/adts.202400637\",\"RegionNum\":4,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://advanced.onlinelibrary.wiley.com/doi/10.1002/adts.202400637","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Electronic Properties of Si and C Substitutional Defects and Porosity in C-Rich and Si-Rich Hydrogenated Roundish SiC Quantum Dots: An Ab-Initio Study
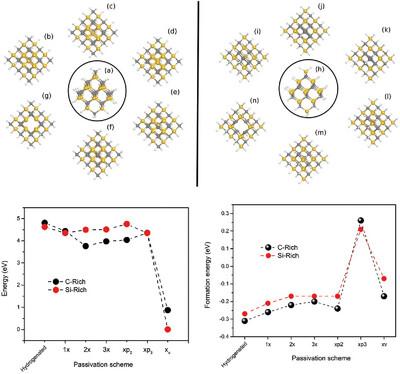
In this study, SiC quantum dots (SiC-QD's) are studied, and some roundish SiC-QD's with the incorporation of defects by removing a carbon or silicon atom are considered. Fourteen configurations are modeled in which the position of the silicon or carbon defect for each configuration is changed, considering that due to the chemical composition, it allows more Si atoms or more C atoms on the QD surface. All calculations are performed using the Density Functional Theory (DFT) methodology. The electronic exchange correlation is treated using the Generalized Gradient Approximation (GGA) with the Revised Perdew–Burke–Ernzerhof (RPBE) functional. The electronic energy levels of each configuration are calculated as well as the partial density of states to know the origin of the energy gap in each quantum dot. The final step is to analyze the energy formation to determine chemical stability.
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
