Mark Zaretckii, Pavel Buslaev, Igor Kozlovskii, Alexander Morozov, Petr Popov
{"title":"利用大型语言模型接近最佳 pH 值酶预测。","authors":"Mark Zaretckii, Pavel Buslaev, Igor Kozlovskii, Alexander Morozov, Petr Popov","doi":"10.1021/acssynbio.4c00465","DOIUrl":null,"url":null,"abstract":"<p><p>Enzymes are widely used in biotechnology due to their ability to catalyze chemical reactions: food making, laundry, pharmaceutics, textile, brewing─all these areas benefit from utilizing various enzymes. Proton concentration (pH) is one of the key factors that define the enzyme functioning and efficiency. Usually there is only a narrow range of pH values where the enzyme is active. This is a common problem in biotechnology to design an enzyme with optimal activity in a given pH range. A large part of this task can be completed <i>in silico</i>, by predicting the optimal pH of designed candidates. The success of such computational methods critically depends on the available data. In this study, we developed a language-model-based approach to predict the optimal pH range from the enzyme sequence. We used different splitting strategies based on sequence similarity, protein family annotation, and enzyme classification to validate the robustness of the proposed approach. The derived machine-learning models demonstrated high accuracy across proteins from different protein families and proteins with lower sequence similarities compared with the training set. The proposed method is fast enough for the high-throughput virtual exploration of protein space for the search for sequences with desired optimal pH levels.</p>","PeriodicalId":26,"journal":{"name":"ACS Synthetic Biology","volume":" ","pages":"3013-3021"},"PeriodicalIF":3.9000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11421216/pdf/","citationCount":"0","resultStr":"{\"title\":\"Approaching Optimal pH Enzyme Prediction with Large Language Models.\",\"authors\":\"Mark Zaretckii, Pavel Buslaev, Igor Kozlovskii, Alexander Morozov, Petr Popov\",\"doi\":\"10.1021/acssynbio.4c00465\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Enzymes are widely used in biotechnology due to their ability to catalyze chemical reactions: food making, laundry, pharmaceutics, textile, brewing─all these areas benefit from utilizing various enzymes. Proton concentration (pH) is one of the key factors that define the enzyme functioning and efficiency. Usually there is only a narrow range of pH values where the enzyme is active. This is a common problem in biotechnology to design an enzyme with optimal activity in a given pH range. A large part of this task can be completed <i>in silico</i>, by predicting the optimal pH of designed candidates. The success of such computational methods critically depends on the available data. In this study, we developed a language-model-based approach to predict the optimal pH range from the enzyme sequence. We used different splitting strategies based on sequence similarity, protein family annotation, and enzyme classification to validate the robustness of the proposed approach. The derived machine-learning models demonstrated high accuracy across proteins from different protein families and proteins with lower sequence similarities compared with the training set. The proposed method is fast enough for the high-throughput virtual exploration of protein space for the search for sequences with desired optimal pH levels.</p>\",\"PeriodicalId\":26,\"journal\":{\"name\":\"ACS Synthetic Biology\",\"volume\":\" \",\"pages\":\"3013-3021\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-09-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11421216/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Synthetic Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1021/acssynbio.4c00465\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Synthetic Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1021/acssynbio.4c00465","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/28 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Approaching Optimal pH Enzyme Prediction with Large Language Models.
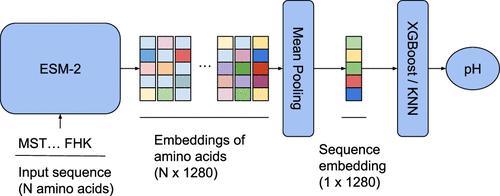
Enzymes are widely used in biotechnology due to their ability to catalyze chemical reactions: food making, laundry, pharmaceutics, textile, brewing─all these areas benefit from utilizing various enzymes. Proton concentration (pH) is one of the key factors that define the enzyme functioning and efficiency. Usually there is only a narrow range of pH values where the enzyme is active. This is a common problem in biotechnology to design an enzyme with optimal activity in a given pH range. A large part of this task can be completed in silico, by predicting the optimal pH of designed candidates. The success of such computational methods critically depends on the available data. In this study, we developed a language-model-based approach to predict the optimal pH range from the enzyme sequence. We used different splitting strategies based on sequence similarity, protein family annotation, and enzyme classification to validate the robustness of the proposed approach. The derived machine-learning models demonstrated high accuracy across proteins from different protein families and proteins with lower sequence similarities compared with the training set. The proposed method is fast enough for the high-throughput virtual exploration of protein space for the search for sequences with desired optimal pH levels.
期刊介绍:
The journal is particularly interested in studies on the design and synthesis of new genetic circuits and gene products; computational methods in the design of systems; and integrative applied approaches to understanding disease and metabolism.
Topics may include, but are not limited to:
Design and optimization of genetic systems
Genetic circuit design and their principles for their organization into programs
Computational methods to aid the design of genetic systems
Experimental methods to quantify genetic parts, circuits, and metabolic fluxes
Genetic parts libraries: their creation, analysis, and ontological representation
Protein engineering including computational design
Metabolic engineering and cellular manufacturing, including biomass conversion
Natural product access, engineering, and production
Creative and innovative applications of cellular programming
Medical applications, tissue engineering, and the programming of therapeutic cells
Minimal cell design and construction
Genomics and genome replacement strategies
Viral engineering
Automated and robotic assembly platforms for synthetic biology
DNA synthesis methodologies
Metagenomics and synthetic metagenomic analysis
Bioinformatics applied to gene discovery, chemoinformatics, and pathway construction
Gene optimization
Methods for genome-scale measurements of transcription and metabolomics
Systems biology and methods to integrate multiple data sources
in vitro and cell-free synthetic biology and molecular programming
Nucleic acid engineering.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
