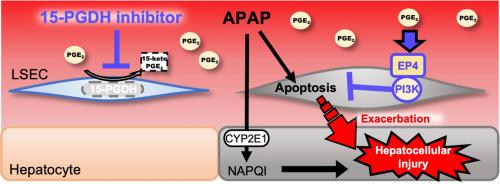
{"title":"抑制 15-前列腺素脱氢酶可通过抑制肝内皮细胞凋亡减轻对乙酰氨基酚诱发的肝损伤","authors":"Hiroaki Shimada , Akito Yokotobi , Nonoka Yamamoto , Mao Takada , Atsushi Kawase , Takeo Nakanishi , Masahiro Iwaki","doi":"10.1016/j.plefa.2024.102640","DOIUrl":null,"url":null,"abstract":"<div><p>Hepatic microvascular disruption caused by injury to liver sinusoidal endothelial cells (LSECs) is an aggravating factor for drug-induced liver injury (DILI). It is suggested that prostaglandin E<sub>2</sub> (PGE<sub>2</sub>) may be able to attenuate LSEC injury. However, it is also known that 15-keto PGE<sub>2</sub>, a metabolite of PGE<sub>2</sub> produced by 15-prostaglandin dehydrogenase (15-PGDH) that is not a ligand of PGE<sub>2</sub> receptors, suppresses inflammatory acute liver injury as a ligand of peroxisome proliferator-activated receptor γ. In this study, we aimed to understand whether 15-PGDH activity is essential for preventing DILI by suppressing hepatic microvascular disruption in a mouse model of acetaminophen (APAP)-induced liver injury. To inhibit 15-PGDH activity prior to APAP-induced LSEC injury, we administered the 15-PGDH inhibitor, SW033291, 1 h before and 3 h after APAP treatment. We observed that LSEC injury preceded hepatocellular injury in APAP administered mice. Hepatic endogenous PGE<sub>2</sub> levels did not increase up till the initiation of LSEC injury but rather increased after hepatocellular injury. Moreover, hepatic 15-PGDH activity was downregulated in APAP-induced liver injury. The inhibition of 15-PGDH attenuated LSEC injury and subsequently hepatic injury by inhibiting apoptosis in APAP administered mice. Our <em>in vitro</em> studies also suggested that PGE<sub>2</sub> inhibited APAP-induced apoptosis via the EP4/PI3K pathway in endothelial cells. Therefore, a decrease in 15-PGDH activity would be beneficial for preventing APAP-induced liver injury by attenuating LSEC injury.</p></div>","PeriodicalId":94179,"journal":{"name":"Prostaglandins, leukotrienes, and essential fatty acids","volume":"202 ","pages":"Article 102640"},"PeriodicalIF":3.2000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Inhibition of 15-prostaglandin dehydrogenase attenuates acetaminophen-induced liver injury via suppression of apoptosis in liver endothelial cells\",\"authors\":\"Hiroaki Shimada , Akito Yokotobi , Nonoka Yamamoto , Mao Takada , Atsushi Kawase , Takeo Nakanishi , Masahiro Iwaki\",\"doi\":\"10.1016/j.plefa.2024.102640\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Hepatic microvascular disruption caused by injury to liver sinusoidal endothelial cells (LSECs) is an aggravating factor for drug-induced liver injury (DILI). It is suggested that prostaglandin E<sub>2</sub> (PGE<sub>2</sub>) may be able to attenuate LSEC injury. However, it is also known that 15-keto PGE<sub>2</sub>, a metabolite of PGE<sub>2</sub> produced by 15-prostaglandin dehydrogenase (15-PGDH) that is not a ligand of PGE<sub>2</sub> receptors, suppresses inflammatory acute liver injury as a ligand of peroxisome proliferator-activated receptor γ. In this study, we aimed to understand whether 15-PGDH activity is essential for preventing DILI by suppressing hepatic microvascular disruption in a mouse model of acetaminophen (APAP)-induced liver injury. To inhibit 15-PGDH activity prior to APAP-induced LSEC injury, we administered the 15-PGDH inhibitor, SW033291, 1 h before and 3 h after APAP treatment. We observed that LSEC injury preceded hepatocellular injury in APAP administered mice. Hepatic endogenous PGE<sub>2</sub> levels did not increase up till the initiation of LSEC injury but rather increased after hepatocellular injury. Moreover, hepatic 15-PGDH activity was downregulated in APAP-induced liver injury. The inhibition of 15-PGDH attenuated LSEC injury and subsequently hepatic injury by inhibiting apoptosis in APAP administered mice. Our <em>in vitro</em> studies also suggested that PGE<sub>2</sub> inhibited APAP-induced apoptosis via the EP4/PI3K pathway in endothelial cells. Therefore, a decrease in 15-PGDH activity would be beneficial for preventing APAP-induced liver injury by attenuating LSEC injury.</p></div>\",\"PeriodicalId\":94179,\"journal\":{\"name\":\"Prostaglandins, leukotrienes, and essential fatty acids\",\"volume\":\"202 \",\"pages\":\"Article 102640\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Prostaglandins, leukotrienes, and essential fatty acids\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0952327824000346\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Prostaglandins, leukotrienes, and essential fatty acids","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0952327824000346","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Inhibition of 15-prostaglandin dehydrogenase attenuates acetaminophen-induced liver injury via suppression of apoptosis in liver endothelial cells
Hepatic microvascular disruption caused by injury to liver sinusoidal endothelial cells (LSECs) is an aggravating factor for drug-induced liver injury (DILI). It is suggested that prostaglandin E2 (PGE2) may be able to attenuate LSEC injury. However, it is also known that 15-keto PGE2, a metabolite of PGE2 produced by 15-prostaglandin dehydrogenase (15-PGDH) that is not a ligand of PGE2 receptors, suppresses inflammatory acute liver injury as a ligand of peroxisome proliferator-activated receptor γ. In this study, we aimed to understand whether 15-PGDH activity is essential for preventing DILI by suppressing hepatic microvascular disruption in a mouse model of acetaminophen (APAP)-induced liver injury. To inhibit 15-PGDH activity prior to APAP-induced LSEC injury, we administered the 15-PGDH inhibitor, SW033291, 1 h before and 3 h after APAP treatment. We observed that LSEC injury preceded hepatocellular injury in APAP administered mice. Hepatic endogenous PGE2 levels did not increase up till the initiation of LSEC injury but rather increased after hepatocellular injury. Moreover, hepatic 15-PGDH activity was downregulated in APAP-induced liver injury. The inhibition of 15-PGDH attenuated LSEC injury and subsequently hepatic injury by inhibiting apoptosis in APAP administered mice. Our in vitro studies also suggested that PGE2 inhibited APAP-induced apoptosis via the EP4/PI3K pathway in endothelial cells. Therefore, a decrease in 15-PGDH activity would be beneficial for preventing APAP-induced liver injury by attenuating LSEC injury.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
