Fangjia Fu , Zhongye Wang , Benkung Hong , Kang Liao , Wei Li
{"title":"用经典和量子力学计算预测小槽粘合剂的结构和光谱特性","authors":"Fangjia Fu , Zhongye Wang , Benkung Hong , Kang Liao , Wei Li","doi":"10.1016/j.cplett.2024.141610","DOIUrl":null,"url":null,"abstract":"<div><p>Regulated DNA-binding ligands can be employed to design fluorescent probes and new drugs for promising chemical and biological applications. However, predicting the binding affinities of DNA ligands based on energy and geometry remains a challenge. In this work, combined molecular docking, molecular dynamics (MD) simulation, and generalized energy-based fragmentation (GEBF) approaches were performed to predict the three binding complexes between DNA and fluorescent or drug molecules. The GEBF binding energy calculation finds that the predicted optimal complexes would agree with the experimental ones in a minor-groove binder, which is more accurate and effective than molecular mechanics and semi-empirical quantum mechanics. The binding site of the water molecule around DAPI-DNA, which forms hydrogen bond networks with the DAPI ligand, can also be predicted with the GEBF method. The result show that the predicted absorption spectra are closely match the experimental measures when considering the solvent, electrostatic, and polarization interactions. Thus, the theoretical calculations using docking, MD simulations, and GEBF methods can be applied to predict the binding modes and sites of DNA ligands, suggesting that it is helpful for virtual screening and designing new small ligands to recognize DNA with various applications.</p></div>","PeriodicalId":273,"journal":{"name":"Chemical Physics Letters","volume":"856 ","pages":"Article 141610"},"PeriodicalIF":3.1000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Predicting structural and spectral properties in a Minor-Groove binder with classical and quantum mechanical calculations\",\"authors\":\"Fangjia Fu , Zhongye Wang , Benkung Hong , Kang Liao , Wei Li\",\"doi\":\"10.1016/j.cplett.2024.141610\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Regulated DNA-binding ligands can be employed to design fluorescent probes and new drugs for promising chemical and biological applications. However, predicting the binding affinities of DNA ligands based on energy and geometry remains a challenge. In this work, combined molecular docking, molecular dynamics (MD) simulation, and generalized energy-based fragmentation (GEBF) approaches were performed to predict the three binding complexes between DNA and fluorescent or drug molecules. The GEBF binding energy calculation finds that the predicted optimal complexes would agree with the experimental ones in a minor-groove binder, which is more accurate and effective than molecular mechanics and semi-empirical quantum mechanics. The binding site of the water molecule around DAPI-DNA, which forms hydrogen bond networks with the DAPI ligand, can also be predicted with the GEBF method. The result show that the predicted absorption spectra are closely match the experimental measures when considering the solvent, electrostatic, and polarization interactions. Thus, the theoretical calculations using docking, MD simulations, and GEBF methods can be applied to predict the binding modes and sites of DNA ligands, suggesting that it is helpful for virtual screening and designing new small ligands to recognize DNA with various applications.</p></div>\",\"PeriodicalId\":273,\"journal\":{\"name\":\"Chemical Physics Letters\",\"volume\":\"856 \",\"pages\":\"Article 141610\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Physics Letters\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0009261424005529\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics Letters","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0009261424005529","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/5 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
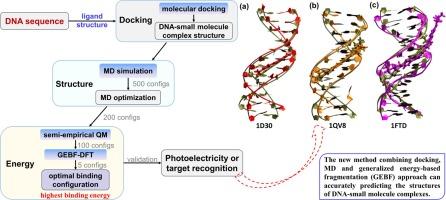
调控 DNA 结合配体可用于设计荧光探针和新药,在化学和生物领域应用前景广阔。然而,根据能量和几何形状预测 DNA 配体的结合亲和力仍然是一项挑战。在这项工作中,研究人员结合分子对接、分子动力学(MD)模拟和基于能量的广义破碎(GEBF)方法,预测了 DNA 与荧光分子或药物分子之间的三种结合复合物。GEBF 结合能计算发现,预测的最佳复合物与实验中的小凹槽粘合剂一致,比分子力学和半经验量子力学更准确有效。水分子在 DAPI-DNA 周围与 DAPI 配体形成氢键网络的结合位点也可以用 GEBF 方法预测。结果表明,在考虑溶剂、静电和极化相互作用的情况下,预测的吸收光谱与实验结果非常吻合。因此,利用对接、MD 模拟和 GEBF 方法进行的理论计算可用于预测 DNA 配体的结合模式和位点,有助于虚拟筛选和设计新的小配体来识别 DNA,并具有多种应用价值。
Predicting structural and spectral properties in a Minor-Groove binder with classical and quantum mechanical calculations
Regulated DNA-binding ligands can be employed to design fluorescent probes and new drugs for promising chemical and biological applications. However, predicting the binding affinities of DNA ligands based on energy and geometry remains a challenge. In this work, combined molecular docking, molecular dynamics (MD) simulation, and generalized energy-based fragmentation (GEBF) approaches were performed to predict the three binding complexes between DNA and fluorescent or drug molecules. The GEBF binding energy calculation finds that the predicted optimal complexes would agree with the experimental ones in a minor-groove binder, which is more accurate and effective than molecular mechanics and semi-empirical quantum mechanics. The binding site of the water molecule around DAPI-DNA, which forms hydrogen bond networks with the DAPI ligand, can also be predicted with the GEBF method. The result show that the predicted absorption spectra are closely match the experimental measures when considering the solvent, electrostatic, and polarization interactions. Thus, the theoretical calculations using docking, MD simulations, and GEBF methods can be applied to predict the binding modes and sites of DNA ligands, suggesting that it is helpful for virtual screening and designing new small ligands to recognize DNA with various applications.
期刊介绍:
Chemical Physics Letters has an open access mirror journal, Chemical Physics Letters: X, sharing the same aims and scope, editorial team, submission system and rigorous peer review.
Chemical Physics Letters publishes brief reports on molecules, interfaces, condensed phases, nanomaterials and nanostructures, polymers, biomolecular systems, and energy conversion and storage.
Criteria for publication are quality, urgency and impact. Further, experimental results reported in the journal have direct relevance for theory, and theoretical developments or non-routine computations relate directly to experiment. Manuscripts must satisfy these criteria and should not be minor extensions of previous work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
