Shibang Lu , Hu Jin , Tiantian Nong , Dongxiao Li , Kang Long , Yanjun Chen , Yan Li , Hao Xing , Tingcai Pan , Songqing He , Keqing Jiang , Fudi Zhong
{"title":"肝细胞衍生的Fetuin-A通过抑制TLR4的自噬-溶酶体降解和M2巨噬细胞极化,促进小鼠酒精相关性肝病的发生","authors":"Shibang Lu , Hu Jin , Tiantian Nong , Dongxiao Li , Kang Long , Yanjun Chen , Yan Li , Hao Xing , Tingcai Pan , Songqing He , Keqing Jiang , Fudi Zhong","doi":"10.1016/j.freeradbiomed.2024.09.011","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Alcohol-associated liver disease (ALD) is one of the most common chronic liver diseases worldwide. Fetuin-A (FetA) is a plasma glycoprotein closely related to fat accumulation in the liver. However, the role of FetA in ALD remains unclear.</p></div><div><h3>Methods</h3><p>Both National Institute on Alcohol Abuse and Alcoholism (NIAAA) model and ethanol (EtOH) treated cell were used in this study. The effect of FetA deficiency on the progression of ALD was analyzed and the underlying mechanism was explored.</p></div><div><h3>Results</h3><p>The expression of FetA was upregulated in the liver tissues of ethanol-fed mice and ALD patients, as well as in AML12 cells treated with ethanol. FetA deletion reduced hepatic steatosis, oxidative stress, and inflammation in ALD mice. Interestingly, the absence of FetA led to a reduction of TLR4 protein level in liver tissue of EtOH-fed mice, without a corresponding change of its mRNA level. Conversely, the administration of recombinant FetA elevated TLR4 protein level in ethanol-treated RAW264.7 cells. FetA knockout significantly impeded the polarization of M1 macrophage <em>in vivo</em> or <em>in vitro</em>. Mechanistically, FetA deficiency drived the autophagy-lysosomal degradation of TLR4, subsequently inhibiting the activation of NF-kB/NLRP3 inflammasome pathway. Furthermore, knockdown of FetA using an adeno-associated virus 8 (AAV8)-shRNA can effectively prevent the progression of ALD in mice.</p></div><div><h3>Conclusion</h3><p>Our results indicate that inhibition of FetA reverses the progression of ALD in mice, implying that FetA can serve as a therapeutic target for the treatment of ALD.</p></div>","PeriodicalId":12407,"journal":{"name":"Free Radical Biology and Medicine","volume":"224 ","pages":"Pages 506-520"},"PeriodicalIF":8.2000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0891584924006580/pdfft?md5=b85a2923279fac8b05598e10a48d247d&pid=1-s2.0-S0891584924006580-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Hepatocyte-derived Fetuin-A promotes alcohol-associated liver disease in mice by inhibiting autophagy-lysosome degradation of TLR4 and M2 macrophage polarization\",\"authors\":\"Shibang Lu , Hu Jin , Tiantian Nong , Dongxiao Li , Kang Long , Yanjun Chen , Yan Li , Hao Xing , Tingcai Pan , Songqing He , Keqing Jiang , Fudi Zhong\",\"doi\":\"10.1016/j.freeradbiomed.2024.09.011\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>Alcohol-associated liver disease (ALD) is one of the most common chronic liver diseases worldwide. Fetuin-A (FetA) is a plasma glycoprotein closely related to fat accumulation in the liver. However, the role of FetA in ALD remains unclear.</p></div><div><h3>Methods</h3><p>Both National Institute on Alcohol Abuse and Alcoholism (NIAAA) model and ethanol (EtOH) treated cell were used in this study. The effect of FetA deficiency on the progression of ALD was analyzed and the underlying mechanism was explored.</p></div><div><h3>Results</h3><p>The expression of FetA was upregulated in the liver tissues of ethanol-fed mice and ALD patients, as well as in AML12 cells treated with ethanol. FetA deletion reduced hepatic steatosis, oxidative stress, and inflammation in ALD mice. Interestingly, the absence of FetA led to a reduction of TLR4 protein level in liver tissue of EtOH-fed mice, without a corresponding change of its mRNA level. Conversely, the administration of recombinant FetA elevated TLR4 protein level in ethanol-treated RAW264.7 cells. FetA knockout significantly impeded the polarization of M1 macrophage <em>in vivo</em> or <em>in vitro</em>. Mechanistically, FetA deficiency drived the autophagy-lysosomal degradation of TLR4, subsequently inhibiting the activation of NF-kB/NLRP3 inflammasome pathway. Furthermore, knockdown of FetA using an adeno-associated virus 8 (AAV8)-shRNA can effectively prevent the progression of ALD in mice.</p></div><div><h3>Conclusion</h3><p>Our results indicate that inhibition of FetA reverses the progression of ALD in mice, implying that FetA can serve as a therapeutic target for the treatment of ALD.</p></div>\",\"PeriodicalId\":12407,\"journal\":{\"name\":\"Free Radical Biology and Medicine\",\"volume\":\"224 \",\"pages\":\"Pages 506-520\"},\"PeriodicalIF\":8.2000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0891584924006580/pdfft?md5=b85a2923279fac8b05598e10a48d247d&pid=1-s2.0-S0891584924006580-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Free Radical Biology and Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0891584924006580\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Free Radical Biology and Medicine","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0891584924006580","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/12 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Hepatocyte-derived Fetuin-A promotes alcohol-associated liver disease in mice by inhibiting autophagy-lysosome degradation of TLR4 and M2 macrophage polarization
Background
Alcohol-associated liver disease (ALD) is one of the most common chronic liver diseases worldwide. Fetuin-A (FetA) is a plasma glycoprotein closely related to fat accumulation in the liver. However, the role of FetA in ALD remains unclear.
Methods
Both National Institute on Alcohol Abuse and Alcoholism (NIAAA) model and ethanol (EtOH) treated cell were used in this study. The effect of FetA deficiency on the progression of ALD was analyzed and the underlying mechanism was explored.
Results
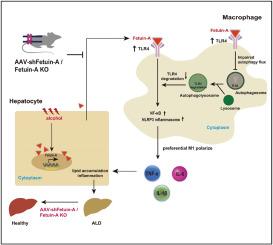
The expression of FetA was upregulated in the liver tissues of ethanol-fed mice and ALD patients, as well as in AML12 cells treated with ethanol. FetA deletion reduced hepatic steatosis, oxidative stress, and inflammation in ALD mice. Interestingly, the absence of FetA led to a reduction of TLR4 protein level in liver tissue of EtOH-fed mice, without a corresponding change of its mRNA level. Conversely, the administration of recombinant FetA elevated TLR4 protein level in ethanol-treated RAW264.7 cells. FetA knockout significantly impeded the polarization of M1 macrophage in vivo or in vitro. Mechanistically, FetA deficiency drived the autophagy-lysosomal degradation of TLR4, subsequently inhibiting the activation of NF-kB/NLRP3 inflammasome pathway. Furthermore, knockdown of FetA using an adeno-associated virus 8 (AAV8)-shRNA can effectively prevent the progression of ALD in mice.
Conclusion
Our results indicate that inhibition of FetA reverses the progression of ALD in mice, implying that FetA can serve as a therapeutic target for the treatment of ALD.
期刊介绍:
Free Radical Biology and Medicine is a leading journal in the field of redox biology, which is the study of the role of reactive oxygen species (ROS) and other oxidizing agents in biological systems. The journal serves as a premier forum for publishing innovative and groundbreaking research that explores the redox biology of health and disease, covering a wide range of topics and disciplines. Free Radical Biology and Medicine also commissions Special Issues that highlight recent advances in both basic and clinical research, with a particular emphasis on the mechanisms underlying altered metabolism and redox signaling. These Special Issues aim to provide a focused platform for the latest research in the field, fostering collaboration and knowledge exchange among researchers and clinicians.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
