Pacharaporn Krawmanee, M. Paul Gleeson, Duangkamol Gleeson
{"title":"Ru 介导的 N-芳基硫脲制备苯并噻唑的计算研究:阐明反应机理和不同底物反应活性的起源","authors":"Pacharaporn Krawmanee, M. Paul Gleeson, Duangkamol Gleeson","doi":"10.1002/qua.27480","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Synthesis of novel benzothiazoles via intramolecular C<span></span>S bond formation reactions is increasingly being explored since they have been found in a wide range of natural products and pharmaceutical agents. Sharma et al. reported the ruthenium-catalyzed preparation of novel benzothiazole derivatives from <i>N</i>-arylthiourea precursors, with a range of reaction yields and selectivity being observed. We have employed a density functional theory-based computational model to investigate the reaction mechanism leading to the benzothiazole product and help uncover the origin of the differing experimental yields and substrate specificities. We proposed a modified mechanistic scheme where the rate-determining step to be the synchronized breaking of the peroxide bond of the oxidizing agent with the concomitant proton-coupled electron transfer from the haloarene urea and a Ru-bound water molecule, not electrophilic Ru<span></span>C bond activation. Evidence for this being the rate-determining step is (a) the barrier is consistent with a lack of kinetic isotope effects associated with the <i>ortho</i>-H atom and (b) the computed rate-determining barriers for 10 <i>N</i>-arylthiourea substrates show good correlation with the observed yield.</p>\n </div>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 19","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational Investigation of the Ru-Mediated Preparation of Benzothiazoles From N-Arylthioureas: Elucidation of the Reaction Mechanism and the Origin of Differing Substrate Reactivity\",\"authors\":\"Pacharaporn Krawmanee, M. Paul Gleeson, Duangkamol Gleeson\",\"doi\":\"10.1002/qua.27480\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>Synthesis of novel benzothiazoles via intramolecular C<span></span>S bond formation reactions is increasingly being explored since they have been found in a wide range of natural products and pharmaceutical agents. Sharma et al. reported the ruthenium-catalyzed preparation of novel benzothiazole derivatives from <i>N</i>-arylthiourea precursors, with a range of reaction yields and selectivity being observed. We have employed a density functional theory-based computational model to investigate the reaction mechanism leading to the benzothiazole product and help uncover the origin of the differing experimental yields and substrate specificities. We proposed a modified mechanistic scheme where the rate-determining step to be the synchronized breaking of the peroxide bond of the oxidizing agent with the concomitant proton-coupled electron transfer from the haloarene urea and a Ru-bound water molecule, not electrophilic Ru<span></span>C bond activation. Evidence for this being the rate-determining step is (a) the barrier is consistent with a lack of kinetic isotope effects associated with the <i>ortho</i>-H atom and (b) the computed rate-determining barriers for 10 <i>N</i>-arylthiourea substrates show good correlation with the observed yield.</p>\\n </div>\",\"PeriodicalId\":182,\"journal\":{\"name\":\"International Journal of Quantum Chemistry\",\"volume\":\"124 19\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Quantum Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/qua.27480\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27480","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Computational Investigation of the Ru-Mediated Preparation of Benzothiazoles From N-Arylthioureas: Elucidation of the Reaction Mechanism and the Origin of Differing Substrate Reactivity
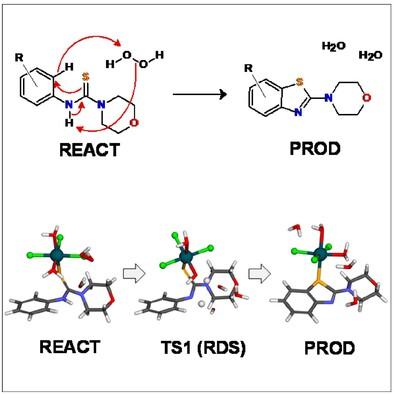
Synthesis of novel benzothiazoles via intramolecular CS bond formation reactions is increasingly being explored since they have been found in a wide range of natural products and pharmaceutical agents. Sharma et al. reported the ruthenium-catalyzed preparation of novel benzothiazole derivatives from N-arylthiourea precursors, with a range of reaction yields and selectivity being observed. We have employed a density functional theory-based computational model to investigate the reaction mechanism leading to the benzothiazole product and help uncover the origin of the differing experimental yields and substrate specificities. We proposed a modified mechanistic scheme where the rate-determining step to be the synchronized breaking of the peroxide bond of the oxidizing agent with the concomitant proton-coupled electron transfer from the haloarene urea and a Ru-bound water molecule, not electrophilic RuC bond activation. Evidence for this being the rate-determining step is (a) the barrier is consistent with a lack of kinetic isotope effects associated with the ortho-H atom and (b) the computed rate-determining barriers for 10 N-arylthiourea substrates show good correlation with the observed yield.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
