{"title":"案例研究:分析症状不明确的肺纤维化患者的 CFTR 基因突变和 SNPs。","authors":"Sahar Yousaf, Sumaira, Iqbal Bano, Atia Rehman, Samra Kousar, Muhammad Usman Ghani, Mariam Shahid","doi":"10.1155/2024/8836342","DOIUrl":null,"url":null,"abstract":"<p><p>Cystic fibrosis (CF) is a genetic monogenic disorder inherited in an autosomal recessive manner, marked by persistent airway infections in the endobronchial region. This condition leads to the gradual development of bronchiectasis and, ultimately, respiratory failure, emerging as the primary cause of mortality in individuals diagnosed with CF. Diagnosis is done depending on the patient's symptoms and lung radiological findings like chest X-rays and CTs. In younger patients and children, diagnosis becomes difficult due to overlapping symptoms with other diseases such as CF which is a rare genetic disease in our population. Diagnosis of CF usually relies on characteristic symptoms, a family history of CF, and an abnormal sweat chloride test, but in children, low sweat production during testing leads to false negative results. In this case report, a suspected patient with ambiguous respiratory symptoms underwent a comprehensive investigation revealing elevated CRP levels, TLC, and characteristic pulmonary manifestations on chest X-ray, suggesting cystic fibrosis. Despite negative sweat chloride tests, the patient was analysed for potential candidate SNPs and was also tested for potential CFTR mutations to rule out CF, genetic analysis confirmed the diagnosis. Genetic testing plays a crucial role in diagnosing cystic fibrosis, even when traditional tests are inconclusive. Specific mutations like Δ508 deletion and rs213950 guide personalized treatment. Consanguinity and family history highlight genetic predisposition, while environmental factors may influence symptom onset. Further research is needed to understand these complexities and improve diagnostic and treatment approaches.</p>","PeriodicalId":9627,"journal":{"name":"Case Reports in Medicine","volume":"2024 ","pages":"8836342"},"PeriodicalIF":0.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11442034/pdf/","citationCount":"0","resultStr":"{\"title\":\"Case Study: Analyzing CFTR Mutations and SNPs in Pulmonary Fibrosis Patients with Unclear Symptoms.\",\"authors\":\"Sahar Yousaf, Sumaira, Iqbal Bano, Atia Rehman, Samra Kousar, Muhammad Usman Ghani, Mariam Shahid\",\"doi\":\"10.1155/2024/8836342\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cystic fibrosis (CF) is a genetic monogenic disorder inherited in an autosomal recessive manner, marked by persistent airway infections in the endobronchial region. This condition leads to the gradual development of bronchiectasis and, ultimately, respiratory failure, emerging as the primary cause of mortality in individuals diagnosed with CF. Diagnosis is done depending on the patient's symptoms and lung radiological findings like chest X-rays and CTs. In younger patients and children, diagnosis becomes difficult due to overlapping symptoms with other diseases such as CF which is a rare genetic disease in our population. Diagnosis of CF usually relies on characteristic symptoms, a family history of CF, and an abnormal sweat chloride test, but in children, low sweat production during testing leads to false negative results. In this case report, a suspected patient with ambiguous respiratory symptoms underwent a comprehensive investigation revealing elevated CRP levels, TLC, and characteristic pulmonary manifestations on chest X-ray, suggesting cystic fibrosis. Despite negative sweat chloride tests, the patient was analysed for potential candidate SNPs and was also tested for potential CFTR mutations to rule out CF, genetic analysis confirmed the diagnosis. Genetic testing plays a crucial role in diagnosing cystic fibrosis, even when traditional tests are inconclusive. Specific mutations like Δ508 deletion and rs213950 guide personalized treatment. Consanguinity and family history highlight genetic predisposition, while environmental factors may influence symptom onset. Further research is needed to understand these complexities and improve diagnostic and treatment approaches.</p>\",\"PeriodicalId\":9627,\"journal\":{\"name\":\"Case Reports in Medicine\",\"volume\":\"2024 \",\"pages\":\"8836342\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11442034/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2024/8836342\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/8836342","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
摘要
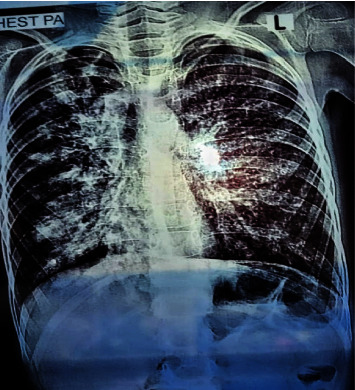
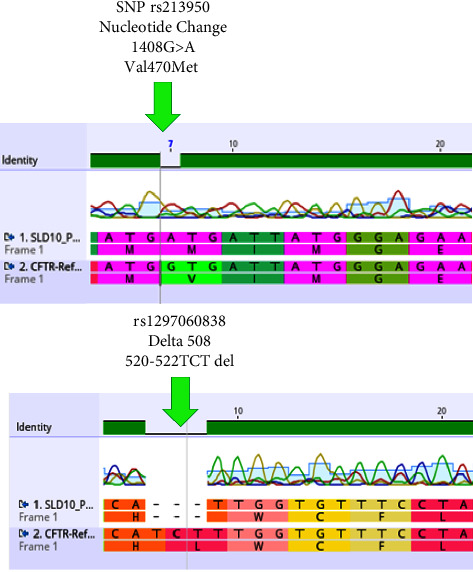
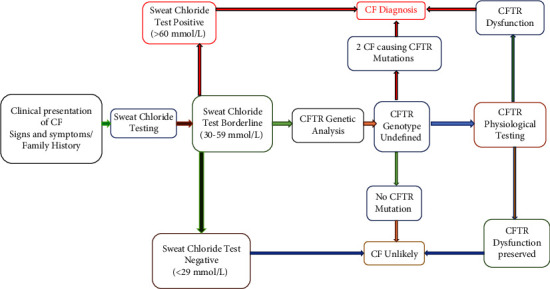
囊性纤维化(CF)是一种常染色体隐性遗传的单基因疾病,以支气管内膜区域持续性气道感染为特征。这种疾病会逐渐导致支气管扩张,最终导致呼吸衰竭,是导致确诊为囊性纤维化患者死亡的主要原因。诊断取决于患者的症状以及胸部 X 光和 CT 等肺部放射学检查结果。对于年龄较小的患者和儿童来说,由于症状与其他疾病(如在我国人群中罕见的遗传性疾病 CF)重叠,诊断变得十分困难。CF的诊断通常依赖于特征性症状、CF家族史和异常的汗液氯化物检测,但在儿童中,检测时汗液分泌过少会导致假阴性结果。在本病例报告中,一名呼吸道症状不明确的疑似患者接受了全面检查,结果显示 CRP 水平升高、TLC 和胸部 X 光片上的特征性肺部表现,提示为囊性纤维化。尽管氯化汗液检测呈阴性,但对患者进行了潜在候选 SNP 分析,并检测了潜在的 CFTR 突变,以排除 CF 的可能性,基因分析证实了诊断。基因检测在诊断囊性纤维化方面起着至关重要的作用,即使传统检测无法得出结论。Δ508缺失和rs213950等特定突变为个性化治疗提供了指导。近亲结婚和家族史突出了遗传易感性,而环境因素可能会影响症状的发作。要了解这些复杂性并改进诊断和治疗方法,还需要进一步的研究。
Case Study: Analyzing CFTR Mutations and SNPs in Pulmonary Fibrosis Patients with Unclear Symptoms.
Cystic fibrosis (CF) is a genetic monogenic disorder inherited in an autosomal recessive manner, marked by persistent airway infections in the endobronchial region. This condition leads to the gradual development of bronchiectasis and, ultimately, respiratory failure, emerging as the primary cause of mortality in individuals diagnosed with CF. Diagnosis is done depending on the patient's symptoms and lung radiological findings like chest X-rays and CTs. In younger patients and children, diagnosis becomes difficult due to overlapping symptoms with other diseases such as CF which is a rare genetic disease in our population. Diagnosis of CF usually relies on characteristic symptoms, a family history of CF, and an abnormal sweat chloride test, but in children, low sweat production during testing leads to false negative results. In this case report, a suspected patient with ambiguous respiratory symptoms underwent a comprehensive investigation revealing elevated CRP levels, TLC, and characteristic pulmonary manifestations on chest X-ray, suggesting cystic fibrosis. Despite negative sweat chloride tests, the patient was analysed for potential candidate SNPs and was also tested for potential CFTR mutations to rule out CF, genetic analysis confirmed the diagnosis. Genetic testing plays a crucial role in diagnosing cystic fibrosis, even when traditional tests are inconclusive. Specific mutations like Δ508 deletion and rs213950 guide personalized treatment. Consanguinity and family history highlight genetic predisposition, while environmental factors may influence symptom onset. Further research is needed to understand these complexities and improve diagnostic and treatment approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
