Darcy Cochran, Panteleimon G. Takis, James L. Alexander, Benjamin H. Mullish, Nick Powell, Julian R. Marchesi and Robert Powers
{"title":"通过核磁共振评估可重复的靶向代谢组学方案","authors":"Darcy Cochran, Panteleimon G. Takis, James L. Alexander, Benjamin H. Mullish, Nick Powell, Julian R. Marchesi and Robert Powers","doi":"10.1039/D4AN01015A","DOIUrl":null,"url":null,"abstract":"<p >Metabolomics aims to study the downstream effects of variables like diet, environment, or disease on a given biological system. However, inconsistencies in sample preparation, data acquisition/processing protocols lead to reproducibility and accuracy concerns. A systematic study was conducted to assess how sample preparation methods and data analysis platforms affect metabolite susceptibility. A targeted panel of 25 metabolites was evaluated in 69 clinical metabolomics samples prepared following three different protocols: intact, ultrafiltration, and protein precipitation. The resulting metabolic profiles were characterized by 1D <small><sup>1</sup></small>H nuclear magnetic resonance (NMR) spectroscopy and analyzed with Chenomx v8.3 and SMolESY software packages. Greater than 90% of the metabolites were extracted more efficiently using protein precipitation than filtration, which aligns with previously reported results. Additionally, analysis of data processing software suggests that metabolite concentrations were overestimated by Chenomx batch-fitting, which only appears reliable for determining relative fold changes rather than absolute quantification. However, an assisted-fit method provided sufficient guidance to achieve accurate results while avoiding a time-consuming fully manual-fitting approach. By combining our results with previous studies, we can now provide a list of 5 common metabolites [2-hydroxybutyrate (2-HB), choline, dimethylamine (DMA), glutamate, lactate] with a high degree of variability in reported fold changes and standard deviations that need careful consideration before being annotated as potential biomarkers. Our results show that sample preparation and data processing package critically impact clinical metabolomics study success. There is a clear need for an increased degree of standardization and harmonization of methods across the metabolomics community to ensure reliable outcomes.</p>","PeriodicalId":63,"journal":{"name":"Analyst","volume":" 22","pages":" 5423-5432"},"PeriodicalIF":3.3000,"publicationDate":"2024-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Evaluating protocols for reproducible targeted metabolomics by NMR†\",\"authors\":\"Darcy Cochran, Panteleimon G. Takis, James L. Alexander, Benjamin H. Mullish, Nick Powell, Julian R. Marchesi and Robert Powers\",\"doi\":\"10.1039/D4AN01015A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Metabolomics aims to study the downstream effects of variables like diet, environment, or disease on a given biological system. However, inconsistencies in sample preparation, data acquisition/processing protocols lead to reproducibility and accuracy concerns. A systematic study was conducted to assess how sample preparation methods and data analysis platforms affect metabolite susceptibility. A targeted panel of 25 metabolites was evaluated in 69 clinical metabolomics samples prepared following three different protocols: intact, ultrafiltration, and protein precipitation. The resulting metabolic profiles were characterized by 1D <small><sup>1</sup></small>H nuclear magnetic resonance (NMR) spectroscopy and analyzed with Chenomx v8.3 and SMolESY software packages. Greater than 90% of the metabolites were extracted more efficiently using protein precipitation than filtration, which aligns with previously reported results. Additionally, analysis of data processing software suggests that metabolite concentrations were overestimated by Chenomx batch-fitting, which only appears reliable for determining relative fold changes rather than absolute quantification. However, an assisted-fit method provided sufficient guidance to achieve accurate results while avoiding a time-consuming fully manual-fitting approach. By combining our results with previous studies, we can now provide a list of 5 common metabolites [2-hydroxybutyrate (2-HB), choline, dimethylamine (DMA), glutamate, lactate] with a high degree of variability in reported fold changes and standard deviations that need careful consideration before being annotated as potential biomarkers. Our results show that sample preparation and data processing package critically impact clinical metabolomics study success. There is a clear need for an increased degree of standardization and harmonization of methods across the metabolomics community to ensure reliable outcomes.</p>\",\"PeriodicalId\":63,\"journal\":{\"name\":\"Analyst\",\"volume\":\" 22\",\"pages\":\" 5423-5432\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-10-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Analyst\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/an/d4an01015a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ANALYTICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Analyst","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/an/d4an01015a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ANALYTICAL","Score":null,"Total":0}
Evaluating protocols for reproducible targeted metabolomics by NMR†
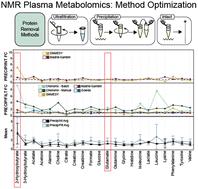
Metabolomics aims to study the downstream effects of variables like diet, environment, or disease on a given biological system. However, inconsistencies in sample preparation, data acquisition/processing protocols lead to reproducibility and accuracy concerns. A systematic study was conducted to assess how sample preparation methods and data analysis platforms affect metabolite susceptibility. A targeted panel of 25 metabolites was evaluated in 69 clinical metabolomics samples prepared following three different protocols: intact, ultrafiltration, and protein precipitation. The resulting metabolic profiles were characterized by 1D 1H nuclear magnetic resonance (NMR) spectroscopy and analyzed with Chenomx v8.3 and SMolESY software packages. Greater than 90% of the metabolites were extracted more efficiently using protein precipitation than filtration, which aligns with previously reported results. Additionally, analysis of data processing software suggests that metabolite concentrations were overestimated by Chenomx batch-fitting, which only appears reliable for determining relative fold changes rather than absolute quantification. However, an assisted-fit method provided sufficient guidance to achieve accurate results while avoiding a time-consuming fully manual-fitting approach. By combining our results with previous studies, we can now provide a list of 5 common metabolites [2-hydroxybutyrate (2-HB), choline, dimethylamine (DMA), glutamate, lactate] with a high degree of variability in reported fold changes and standard deviations that need careful consideration before being annotated as potential biomarkers. Our results show that sample preparation and data processing package critically impact clinical metabolomics study success. There is a clear need for an increased degree of standardization and harmonization of methods across the metabolomics community to ensure reliable outcomes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
