{"title":"Cs2(Ti, Zr, Hf)X6 双卤化物包晶带偏移的第一原理计算","authors":"Yongyut Laosiritaworn, Atchara Punya Jaroenjittichai","doi":"10.1016/j.commatsci.2024.113436","DOIUrl":null,"url":null,"abstract":"<div><div>This study investigates the band offsets among Cs<sub>2</sub>(Ti, Zr, Hf)X<sub>6</sub> double halide perovskites. Valence band offsets (VBO) and conduction band offsets (CBO) were calculated using density functional theory (DFT) with the Perdew–Burke-Ernzerhof (PBE) functional and the hybrid Heyd–Scuseria–Ernzerhof (HSE) functional, employing the supercell technique. This technique offers greater accuracy and reliability by explicitly calculating the potential differences at the interface. A critical factor influencing the results is the contribution of the dipole potential (V<sub>D</sub>), which induces shifts in the VBO and CBO by approximately 0.2 to 0.6 eV relative to values predicted by the electron affinity rule. This discrepancy arises from the inclusion of interface-specific effects, such as charge redistribution and polarization. Additionally, the findings indicate that the energy band alignments among these compounds are type-I within the same group of halides, with nearly identical lattice constants for Zr- and Hf-based compounds due to their similar ionic radii. These results provide valuable insights for the design of heterostructures in electronic applications and highlight the potential of Cs<sub>2</sub>(Ti, Zr, Hf)X<sub>6</sub> compounds as efficient materials for solar cells, light-emitting diodes, and photodetectors.</div></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"246 ","pages":"Article 113436"},"PeriodicalIF":3.7000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The first principle calculations of band offsets among Cs2(Ti, Zr, Hf)X6 double halide perovskites\",\"authors\":\"Yongyut Laosiritaworn, Atchara Punya Jaroenjittichai\",\"doi\":\"10.1016/j.commatsci.2024.113436\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>This study investigates the band offsets among Cs<sub>2</sub>(Ti, Zr, Hf)X<sub>6</sub> double halide perovskites. Valence band offsets (VBO) and conduction band offsets (CBO) were calculated using density functional theory (DFT) with the Perdew–Burke-Ernzerhof (PBE) functional and the hybrid Heyd–Scuseria–Ernzerhof (HSE) functional, employing the supercell technique. This technique offers greater accuracy and reliability by explicitly calculating the potential differences at the interface. A critical factor influencing the results is the contribution of the dipole potential (V<sub>D</sub>), which induces shifts in the VBO and CBO by approximately 0.2 to 0.6 eV relative to values predicted by the electron affinity rule. This discrepancy arises from the inclusion of interface-specific effects, such as charge redistribution and polarization. Additionally, the findings indicate that the energy band alignments among these compounds are type-I within the same group of halides, with nearly identical lattice constants for Zr- and Hf-based compounds due to their similar ionic radii. These results provide valuable insights for the design of heterostructures in electronic applications and highlight the potential of Cs<sub>2</sub>(Ti, Zr, Hf)X<sub>6</sub> compounds as efficient materials for solar cells, light-emitting diodes, and photodetectors.</div></div>\",\"PeriodicalId\":10650,\"journal\":{\"name\":\"Computational Materials Science\",\"volume\":\"246 \",\"pages\":\"Article 113436\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational Materials Science\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0927025624006578\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025624006578","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/4 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
The first principle calculations of band offsets among Cs2(Ti, Zr, Hf)X6 double halide perovskites
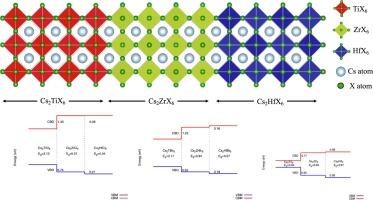
This study investigates the band offsets among Cs2(Ti, Zr, Hf)X6 double halide perovskites. Valence band offsets (VBO) and conduction band offsets (CBO) were calculated using density functional theory (DFT) with the Perdew–Burke-Ernzerhof (PBE) functional and the hybrid Heyd–Scuseria–Ernzerhof (HSE) functional, employing the supercell technique. This technique offers greater accuracy and reliability by explicitly calculating the potential differences at the interface. A critical factor influencing the results is the contribution of the dipole potential (VD), which induces shifts in the VBO and CBO by approximately 0.2 to 0.6 eV relative to values predicted by the electron affinity rule. This discrepancy arises from the inclusion of interface-specific effects, such as charge redistribution and polarization. Additionally, the findings indicate that the energy band alignments among these compounds are type-I within the same group of halides, with nearly identical lattice constants for Zr- and Hf-based compounds due to their similar ionic radii. These results provide valuable insights for the design of heterostructures in electronic applications and highlight the potential of Cs2(Ti, Zr, Hf)X6 compounds as efficient materials for solar cells, light-emitting diodes, and photodetectors.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
