{"title":"基于子结构解释化合物预测的 MolAnchor 方法","authors":"Alec Lamens , Jürgen Bajorath","doi":"10.1016/j.ejmcr.2024.100230","DOIUrl":null,"url":null,"abstract":"<div><div>In medicinal chemistry, the impact of machine learning remains limited if predictions are not understood, which often precludes experimental follow-up. Therefore, chemically intuitive approaches that aid in model understanding and interpretation at the molecular level of detail are sought after. While feature attribution methods quantifying feature importance for model decisions are widely used in many areas, they must typically be combined with visualization techniques, if possible, to render the results accessible from a chemical viewpoint. On the other hand, there are approaches such as counterfactuals that yield closely related chemical structures with different prediction outcomes, providing direct access to structural features that critically influence model decisions. Herein, we introduce another approach designed to rationalize chemical predictions based on molecular structure. Therefore, we adapt principles underlying the anchor concept from explainable artificial intelligence (XAI) and alter them for molecular machine learning. The resulting method, termed MolAnchor, systematically identifies substructures in test compounds that determine property predictions, thus ensuring chemical interpretability. The MolAnchor methodology is made freely available to the medicinal chemistry community as a part of our study.</div></div>","PeriodicalId":12015,"journal":{"name":"European Journal of Medicinal Chemistry Reports","volume":"12 ","pages":"Article 100230"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"MolAnchor method for explaining compound predictions based on substructures\",\"authors\":\"Alec Lamens , Jürgen Bajorath\",\"doi\":\"10.1016/j.ejmcr.2024.100230\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>In medicinal chemistry, the impact of machine learning remains limited if predictions are not understood, which often precludes experimental follow-up. Therefore, chemically intuitive approaches that aid in model understanding and interpretation at the molecular level of detail are sought after. While feature attribution methods quantifying feature importance for model decisions are widely used in many areas, they must typically be combined with visualization techniques, if possible, to render the results accessible from a chemical viewpoint. On the other hand, there are approaches such as counterfactuals that yield closely related chemical structures with different prediction outcomes, providing direct access to structural features that critically influence model decisions. Herein, we introduce another approach designed to rationalize chemical predictions based on molecular structure. Therefore, we adapt principles underlying the anchor concept from explainable artificial intelligence (XAI) and alter them for molecular machine learning. The resulting method, termed MolAnchor, systematically identifies substructures in test compounds that determine property predictions, thus ensuring chemical interpretability. The MolAnchor methodology is made freely available to the medicinal chemistry community as a part of our study.</div></div>\",\"PeriodicalId\":12015,\"journal\":{\"name\":\"European Journal of Medicinal Chemistry Reports\",\"volume\":\"12 \",\"pages\":\"Article 100230\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Medicinal Chemistry Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S277241742400102X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry Reports","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S277241742400102X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/15 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
MolAnchor method for explaining compound predictions based on substructures
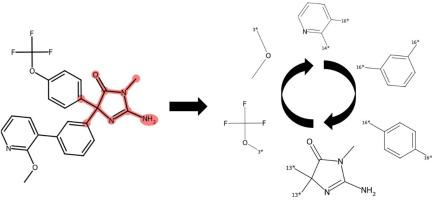
In medicinal chemistry, the impact of machine learning remains limited if predictions are not understood, which often precludes experimental follow-up. Therefore, chemically intuitive approaches that aid in model understanding and interpretation at the molecular level of detail are sought after. While feature attribution methods quantifying feature importance for model decisions are widely used in many areas, they must typically be combined with visualization techniques, if possible, to render the results accessible from a chemical viewpoint. On the other hand, there are approaches such as counterfactuals that yield closely related chemical structures with different prediction outcomes, providing direct access to structural features that critically influence model decisions. Herein, we introduce another approach designed to rationalize chemical predictions based on molecular structure. Therefore, we adapt principles underlying the anchor concept from explainable artificial intelligence (XAI) and alter them for molecular machine learning. The resulting method, termed MolAnchor, systematically identifies substructures in test compounds that determine property predictions, thus ensuring chemical interpretability. The MolAnchor methodology is made freely available to the medicinal chemistry community as a part of our study.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
