Elisabeth Hellec, Flavia Nunes, Charlotte Corporeau, Alexandre Cormier
{"title":"KiNext:用于蛋白激酶鉴定和分类的便携式可扩展工作流程。","authors":"Elisabeth Hellec, Flavia Nunes, Charlotte Corporeau, Alexandre Cormier","doi":"10.1186/s12859-024-05953-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Protein kinases are a diverse superfamily of proteins common to organisms across the tree of life that are typically involved in signal transduction, allowing organisms to sense and respond to biotic or abiotic environmental factors. They have important roles in organismal physiology, including development, reproduction, acclimation to environmental stress, while their dysregulation can lead to disease, including several forms of cancer. Identifying the complement of protein kinases (the kinome) of any organism is useful for understanding its physiological capabilities, limitations and adaptations to environmental stress. The increasing availability of genomes makes it now possible to examine and compare the kinomes across a broad diversity of organisms. Here we present a pipeline respecting the FAIR principles (findable, accessible, interoperable and reusable) that facilitates the search and identification of protein kinases from a predicted proteome, and classifies them according to group of serine/threonine/tyrosine protein kinases present in eukaryotes.</p><p><strong>Results: </strong>KiNext is a Nextflow pipeline that regroups a number of existing bioinformatic tools to search for and classify the protein kinases of an organism in a reproducible manner, starting from a set of amino acid sequences. Conventional eukaryotic protein kinases (ePKs) and atypical protein kinases (aPKs) are identified by using Hidden Markov Models (HMMs) generated from the catalytic domains of kinases. Furthermore, KiNext categorizes ePKs into the eight kinase groups by employing dedicated Hidden Markov Models (HMMs) tailored for each group. The performance of the KiNext pipeline was validated against previously identified kinomes obtained with other tools that were already published for two marine species, the Pacific oyster Crassostrea gigas and the unicellular green alga Ostreoccocus tauri. KiNext outperformed previous results by finding previously unidentified kinases and by attributing a large proportion of previously unclassified kinases to a group in both species. These results demonstrate improvements in kinase identification and classification, all while providing traceability and reproducibility of results in a FAIR pipeline. The default HMM models provided with KiNext are most suitable for eukaryotes, but the pipeline can be easily modified to include HMM models for other taxa of interest.</p><p><strong>Conclusion: </strong>The KiNext pipeline enables efficient and reproducible identification of kinomes based on predicted amino acid sequences (i.e. proteomes). KiNext was designed to be easy to use, automated, portable and scalable.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"25 1","pages":"338"},"PeriodicalIF":4.4000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11515245/pdf/","citationCount":"0","resultStr":"{\"title\":\"KiNext: a portable and scalable workflow for the identification and classification of protein kinases.\",\"authors\":\"Elisabeth Hellec, Flavia Nunes, Charlotte Corporeau, Alexandre Cormier\",\"doi\":\"10.1186/s12859-024-05953-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Protein kinases are a diverse superfamily of proteins common to organisms across the tree of life that are typically involved in signal transduction, allowing organisms to sense and respond to biotic or abiotic environmental factors. They have important roles in organismal physiology, including development, reproduction, acclimation to environmental stress, while their dysregulation can lead to disease, including several forms of cancer. Identifying the complement of protein kinases (the kinome) of any organism is useful for understanding its physiological capabilities, limitations and adaptations to environmental stress. The increasing availability of genomes makes it now possible to examine and compare the kinomes across a broad diversity of organisms. Here we present a pipeline respecting the FAIR principles (findable, accessible, interoperable and reusable) that facilitates the search and identification of protein kinases from a predicted proteome, and classifies them according to group of serine/threonine/tyrosine protein kinases present in eukaryotes.</p><p><strong>Results: </strong>KiNext is a Nextflow pipeline that regroups a number of existing bioinformatic tools to search for and classify the protein kinases of an organism in a reproducible manner, starting from a set of amino acid sequences. Conventional eukaryotic protein kinases (ePKs) and atypical protein kinases (aPKs) are identified by using Hidden Markov Models (HMMs) generated from the catalytic domains of kinases. Furthermore, KiNext categorizes ePKs into the eight kinase groups by employing dedicated Hidden Markov Models (HMMs) tailored for each group. The performance of the KiNext pipeline was validated against previously identified kinomes obtained with other tools that were already published for two marine species, the Pacific oyster Crassostrea gigas and the unicellular green alga Ostreoccocus tauri. KiNext outperformed previous results by finding previously unidentified kinases and by attributing a large proportion of previously unclassified kinases to a group in both species. These results demonstrate improvements in kinase identification and classification, all while providing traceability and reproducibility of results in a FAIR pipeline. The default HMM models provided with KiNext are most suitable for eukaryotes, but the pipeline can be easily modified to include HMM models for other taxa of interest.</p><p><strong>Conclusion: </strong>The KiNext pipeline enables efficient and reproducible identification of kinomes based on predicted amino acid sequences (i.e. proteomes). KiNext was designed to be easy to use, automated, portable and scalable.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"25 1\",\"pages\":\"338\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2024-10-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11515245/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-024-05953-w\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-024-05953-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
KiNext: a portable and scalable workflow for the identification and classification of protein kinases.
Background: Protein kinases are a diverse superfamily of proteins common to organisms across the tree of life that are typically involved in signal transduction, allowing organisms to sense and respond to biotic or abiotic environmental factors. They have important roles in organismal physiology, including development, reproduction, acclimation to environmental stress, while their dysregulation can lead to disease, including several forms of cancer. Identifying the complement of protein kinases (the kinome) of any organism is useful for understanding its physiological capabilities, limitations and adaptations to environmental stress. The increasing availability of genomes makes it now possible to examine and compare the kinomes across a broad diversity of organisms. Here we present a pipeline respecting the FAIR principles (findable, accessible, interoperable and reusable) that facilitates the search and identification of protein kinases from a predicted proteome, and classifies them according to group of serine/threonine/tyrosine protein kinases present in eukaryotes.
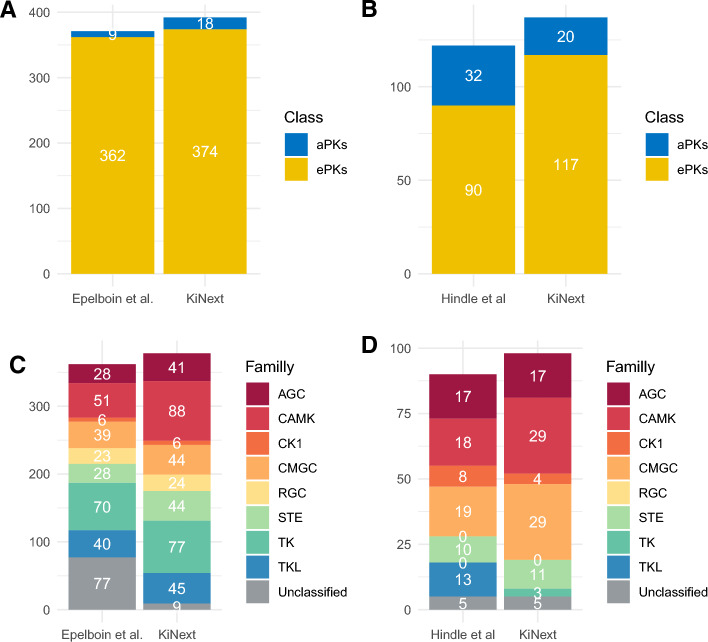
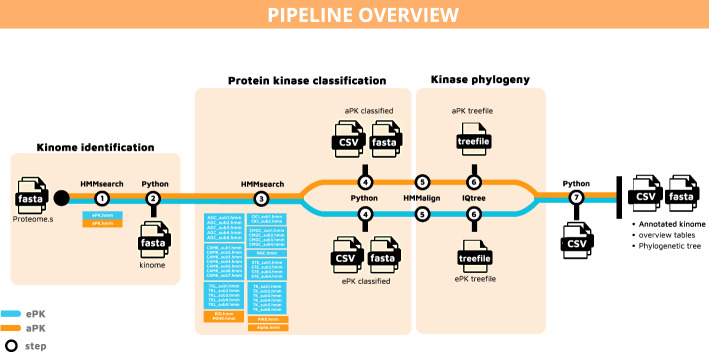
Results: KiNext is a Nextflow pipeline that regroups a number of existing bioinformatic tools to search for and classify the protein kinases of an organism in a reproducible manner, starting from a set of amino acid sequences. Conventional eukaryotic protein kinases (ePKs) and atypical protein kinases (aPKs) are identified by using Hidden Markov Models (HMMs) generated from the catalytic domains of kinases. Furthermore, KiNext categorizes ePKs into the eight kinase groups by employing dedicated Hidden Markov Models (HMMs) tailored for each group. The performance of the KiNext pipeline was validated against previously identified kinomes obtained with other tools that were already published for two marine species, the Pacific oyster Crassostrea gigas and the unicellular green alga Ostreoccocus tauri. KiNext outperformed previous results by finding previously unidentified kinases and by attributing a large proportion of previously unclassified kinases to a group in both species. These results demonstrate improvements in kinase identification and classification, all while providing traceability and reproducibility of results in a FAIR pipeline. The default HMM models provided with KiNext are most suitable for eukaryotes, but the pipeline can be easily modified to include HMM models for other taxa of interest.
Conclusion: The KiNext pipeline enables efficient and reproducible identification of kinomes based on predicted amino acid sequences (i.e. proteomes). KiNext was designed to be easy to use, automated, portable and scalable.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
