Li-Qiang Wang, Yeyang Ma, Mu-Ya Zhang, Han-Ye Yuan, Xiang-Ning Li, Wencheng Xia, Kun Zhao, Xi Huang, Jie Chen, Dan Li, Liangyu Zou, Zhengzhi Wang, Weidong Le, Cong Liu, Yi Liang
{"title":"导致 ALS 的 SOD1 基因突变诱发的淀粉样纤维结构和铁变态反应激活。","authors":"Li-Qiang Wang, Yeyang Ma, Mu-Ya Zhang, Han-Ye Yuan, Xiang-Ning Li, Wencheng Xia, Kun Zhao, Xi Huang, Jie Chen, Dan Li, Liangyu Zou, Zhengzhi Wang, Weidong Le, Cong Liu, Yi Liang","doi":"10.1126/sciadv.ado8499","DOIUrl":null,"url":null,"abstract":"<div >Over 200 genetic mutations in copper-zinc superoxide dismutase (SOD1) have been linked to amyotrophic lateral sclerosis (ALS). Among these, two ALS-causing mutants, histidine-46→arginine (H46R) and glycine-85→arginine (G85R), exhibit a decreased capacity to bind metal ions. Here, we report two cryo–electron microscopy structures of amyloid fibrils formed by H46R and G85R. These mutations lead to the formation of amyloid fibrils with unique structures distinct from those of the native fibril. The core of these fibrils features a serpentine arrangement with seven or eight β strands, secured by a hydrophobic cavity and a salt bridge between arginine-85 and aspartic acid–101 in the G85R fibril. We demonstrate that these mutant fibrils are notably more toxic and capable of promoting the aggregation of wild-type SOD1 more effectively, causing mitochondrial impairment and activating ferroptosis in cell cultures, compared to wild-type SOD1 fibrils. Our study provides insights into the structural mechanisms by which SOD1 mutants aggregate and induce cytotoxicity in ALS.</div>","PeriodicalId":21609,"journal":{"name":"Science Advances","volume":"10 44","pages":""},"PeriodicalIF":12.5000,"publicationDate":"2024-10-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.science.org/doi/reader/10.1126/sciadv.ado8499","citationCount":"0","resultStr":"{\"title\":\"Amyloid fibril structures and ferroptosis activation induced by ALS-causing SOD1 mutations\",\"authors\":\"Li-Qiang Wang, Yeyang Ma, Mu-Ya Zhang, Han-Ye Yuan, Xiang-Ning Li, Wencheng Xia, Kun Zhao, Xi Huang, Jie Chen, Dan Li, Liangyu Zou, Zhengzhi Wang, Weidong Le, Cong Liu, Yi Liang\",\"doi\":\"10.1126/sciadv.ado8499\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div >Over 200 genetic mutations in copper-zinc superoxide dismutase (SOD1) have been linked to amyotrophic lateral sclerosis (ALS). Among these, two ALS-causing mutants, histidine-46→arginine (H46R) and glycine-85→arginine (G85R), exhibit a decreased capacity to bind metal ions. Here, we report two cryo–electron microscopy structures of amyloid fibrils formed by H46R and G85R. These mutations lead to the formation of amyloid fibrils with unique structures distinct from those of the native fibril. The core of these fibrils features a serpentine arrangement with seven or eight β strands, secured by a hydrophobic cavity and a salt bridge between arginine-85 and aspartic acid–101 in the G85R fibril. We demonstrate that these mutant fibrils are notably more toxic and capable of promoting the aggregation of wild-type SOD1 more effectively, causing mitochondrial impairment and activating ferroptosis in cell cultures, compared to wild-type SOD1 fibrils. Our study provides insights into the structural mechanisms by which SOD1 mutants aggregate and induce cytotoxicity in ALS.</div>\",\"PeriodicalId\":21609,\"journal\":{\"name\":\"Science Advances\",\"volume\":\"10 44\",\"pages\":\"\"},\"PeriodicalIF\":12.5000,\"publicationDate\":\"2024-10-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.science.org/doi/reader/10.1126/sciadv.ado8499\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Science Advances\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://www.science.org/doi/10.1126/sciadv.ado8499\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Science Advances","FirstCategoryId":"103","ListUrlMain":"https://www.science.org/doi/10.1126/sciadv.ado8499","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
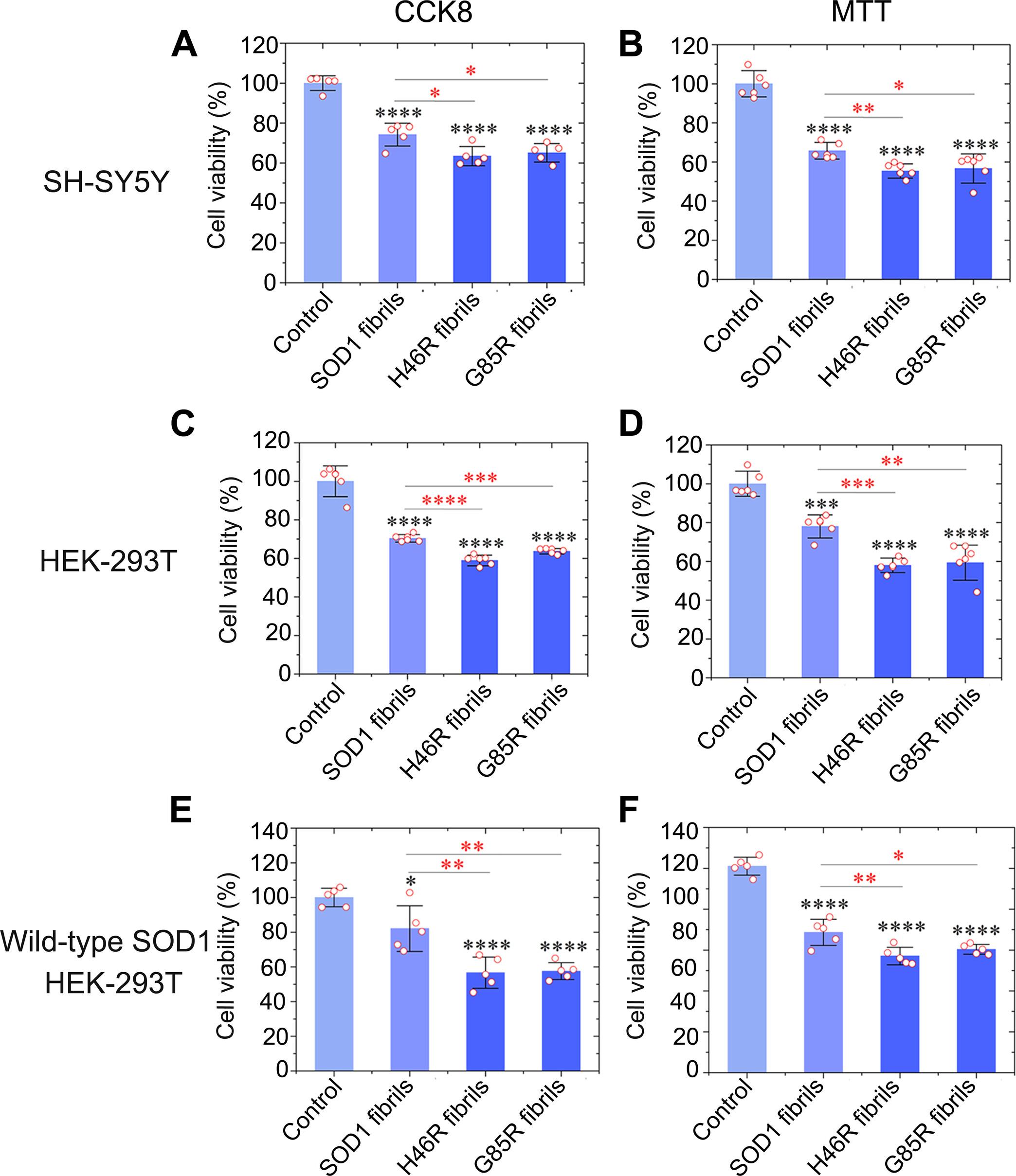
铜锌超氧化物歧化酶(SOD1)中有 200 多种基因突变与肌萎缩性脊髓侧索硬化症(ALS)有关。其中,组氨酸-46→精氨酸(H46R)和甘氨酸-85→精氨酸(G85R)这两种导致 ALS 的突变体表现出结合金属离子的能力下降。在此,我们报告了两种由 H46R 和 G85R 形成的淀粉样纤维的冷冻电镜结构。这些突变导致形成的淀粉样纤维具有不同于原生纤维的独特结构。这些纤丝的核心具有七或八条β链的蛇形排列,由一个疏水空腔和G85R纤丝中精氨酸-85与天冬氨酸-101之间的盐桥固定。我们证明,与野生型 SOD1 纤维相比,这些突变体纤维的毒性明显更强,能更有效地促进野生型 SOD1 的聚集,导致线粒体受损并激活细胞培养物中的铁变态反应。我们的研究深入揭示了 SOD1 突变体在 ALS 中聚集和诱导细胞毒性的结构机制。
Amyloid fibril structures and ferroptosis activation induced by ALS-causing SOD1 mutations
Over 200 genetic mutations in copper-zinc superoxide dismutase (SOD1) have been linked to amyotrophic lateral sclerosis (ALS). Among these, two ALS-causing mutants, histidine-46→arginine (H46R) and glycine-85→arginine (G85R), exhibit a decreased capacity to bind metal ions. Here, we report two cryo–electron microscopy structures of amyloid fibrils formed by H46R and G85R. These mutations lead to the formation of amyloid fibrils with unique structures distinct from those of the native fibril. The core of these fibrils features a serpentine arrangement with seven or eight β strands, secured by a hydrophobic cavity and a salt bridge between arginine-85 and aspartic acid–101 in the G85R fibril. We demonstrate that these mutant fibrils are notably more toxic and capable of promoting the aggregation of wild-type SOD1 more effectively, causing mitochondrial impairment and activating ferroptosis in cell cultures, compared to wild-type SOD1 fibrils. Our study provides insights into the structural mechanisms by which SOD1 mutants aggregate and induce cytotoxicity in ALS.
期刊介绍:
Science Advances, an open-access journal by AAAS, publishes impactful research in diverse scientific areas. It aims for fair, fast, and expert peer review, providing freely accessible research to readers. Led by distinguished scientists, the journal supports AAAS's mission by extending Science magazine's capacity to identify and promote significant advances. Evolving digital publishing technologies play a crucial role in advancing AAAS's global mission for science communication and benefitting humankind.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
