Yang Wang , Sen Hong , Hannah Hudson , Nora Kory , Lisa N. Kinch , Julia Kozlitina , Jonathan C. Cohen , Helen H. Hobbs
{"title":"PNPLA3(148M) 是一种功能增益突变,可通过抑制 ATGL 介导的甘油三酯水解促进肝脂肪变性","authors":"Yang Wang , Sen Hong , Hannah Hudson , Nora Kory , Lisa N. Kinch , Julia Kozlitina , Jonathan C. Cohen , Helen H. Hobbs","doi":"10.1016/j.jhep.2024.10.048","DOIUrl":null,"url":null,"abstract":"<div><h3>Background & Aims</h3><div>PNPLA3(148M) (patatin-like phospholipase domain-containing protein 3) is the most impactful genetic risk factor for steatotic liver disease. A key unresolved issue is whether PNPLA3(148M) confers a loss- or gain-of-function. Here we test the hypothesis that PNPLA3 causes steatosis by sequestering ABHD5 (α/β hydrolase domain-containing protein 5), the cofactor of ATGL (adipose TG lipase), thus limiting mobilization of hepatic triglyceride (TG).</div></div><div><h3>Methods</h3><div>We quantified and compared the physical interactions between ABHD5 and PNPLA3/ATGL in cultured hepatocytes using NanoBiT complementation assays and immunocytochemistry. Recombinant proteins purified from human cells were used to compare TG hydrolytic activities of PNPLA3 and ATGL in the presence or absence of ABHD5. Adenoviruses and adeno-associated viruses were used to express PNPLA3 in liver-specific <em>Atgl</em><sup><em>-/-</em></sup> mice and to express ABHD5 in livers of <em>Pnpla3</em><sup><em>M/M</em></sup> mice, respectively.</div></div><div><h3>Results</h3><div>ABHD5 interacted preferentially with PNPLA3 relative to ATGL in cultured hepatocytes. No differences were seen in the strength of the interactions between ABHD5 with PNPLA3(WT) and PNPLA3(148M). In contrast to prior findings, we found that PNPLA3, like ATGL, is activated by ABHD5 in <em>in vitro</em> assays using purified proteins. PNPLA3(148M)-associated inhibition of TG hydrolysis required that ATGL be expressed and that PNPLA3 be located on lipid droplets. Finally, overexpression of ABHD5 reversed the hepatic steatosis in <em>Pnpla3</em><sup><em>M/M</em></sup> mice.</div></div><div><h3>Conclusions</h3><div>These findings support the premise that PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by accumulating on lipid droplets and inhibiting ATGL-mediated lipolysis in an ABHD5-dependent manner. Our results predict that reducing, rather than increasing, PNPLA3 expression will be the best strategy to treat PNPLA3(148M)-associated steatotic liver disease.</div></div><div><h3>Impact and implications</h3><div>Steatotic liver disease (SLD) is a common complex disorder associated with both environmental and genetic risk factors. PNPLA3(148M) is the most impactful genetic risk factor for SLD and yet its pathogenic mechanism remains controversial. Herein, we provide evidence that PNPLA3(148M) promotes triglyceride (TG) accumulation by sequestering ABHD5, thus limiting its availability to activate ATGL. Although the substitution of methionine for isoleucine reduces the TG hydrolase activity of PNPLA3, the loss of enzymatic function is not directly related to the steatotic effect of the variant. It is the resulting accumulation of PNPLA3 on LDs that confers a gain-of-function by interfering with ATGL-mediated TG hydrolysis. These findings have implications for the design of potential PNPLA3(148M)-based therapies. Reducing, rather than increasing, PNPLA3 levels is predicted to reverse steatosis in susceptible individuals.</div></div>","PeriodicalId":15888,"journal":{"name":"Journal of Hepatology","volume":"82 5","pages":"Pages 871-881"},"PeriodicalIF":33.0000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by inhibiting ATGL-mediated triglyceride hydrolysis\",\"authors\":\"Yang Wang , Sen Hong , Hannah Hudson , Nora Kory , Lisa N. Kinch , Julia Kozlitina , Jonathan C. Cohen , Helen H. Hobbs\",\"doi\":\"10.1016/j.jhep.2024.10.048\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background & Aims</h3><div>PNPLA3(148M) (patatin-like phospholipase domain-containing protein 3) is the most impactful genetic risk factor for steatotic liver disease. A key unresolved issue is whether PNPLA3(148M) confers a loss- or gain-of-function. Here we test the hypothesis that PNPLA3 causes steatosis by sequestering ABHD5 (α/β hydrolase domain-containing protein 5), the cofactor of ATGL (adipose TG lipase), thus limiting mobilization of hepatic triglyceride (TG).</div></div><div><h3>Methods</h3><div>We quantified and compared the physical interactions between ABHD5 and PNPLA3/ATGL in cultured hepatocytes using NanoBiT complementation assays and immunocytochemistry. Recombinant proteins purified from human cells were used to compare TG hydrolytic activities of PNPLA3 and ATGL in the presence or absence of ABHD5. Adenoviruses and adeno-associated viruses were used to express PNPLA3 in liver-specific <em>Atgl</em><sup><em>-/-</em></sup> mice and to express ABHD5 in livers of <em>Pnpla3</em><sup><em>M/M</em></sup> mice, respectively.</div></div><div><h3>Results</h3><div>ABHD5 interacted preferentially with PNPLA3 relative to ATGL in cultured hepatocytes. No differences were seen in the strength of the interactions between ABHD5 with PNPLA3(WT) and PNPLA3(148M). In contrast to prior findings, we found that PNPLA3, like ATGL, is activated by ABHD5 in <em>in vitro</em> assays using purified proteins. PNPLA3(148M)-associated inhibition of TG hydrolysis required that ATGL be expressed and that PNPLA3 be located on lipid droplets. Finally, overexpression of ABHD5 reversed the hepatic steatosis in <em>Pnpla3</em><sup><em>M/M</em></sup> mice.</div></div><div><h3>Conclusions</h3><div>These findings support the premise that PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by accumulating on lipid droplets and inhibiting ATGL-mediated lipolysis in an ABHD5-dependent manner. Our results predict that reducing, rather than increasing, PNPLA3 expression will be the best strategy to treat PNPLA3(148M)-associated steatotic liver disease.</div></div><div><h3>Impact and implications</h3><div>Steatotic liver disease (SLD) is a common complex disorder associated with both environmental and genetic risk factors. PNPLA3(148M) is the most impactful genetic risk factor for SLD and yet its pathogenic mechanism remains controversial. Herein, we provide evidence that PNPLA3(148M) promotes triglyceride (TG) accumulation by sequestering ABHD5, thus limiting its availability to activate ATGL. Although the substitution of methionine for isoleucine reduces the TG hydrolase activity of PNPLA3, the loss of enzymatic function is not directly related to the steatotic effect of the variant. It is the resulting accumulation of PNPLA3 on LDs that confers a gain-of-function by interfering with ATGL-mediated TG hydrolysis. These findings have implications for the design of potential PNPLA3(148M)-based therapies. Reducing, rather than increasing, PNPLA3 levels is predicted to reverse steatosis in susceptible individuals.</div></div>\",\"PeriodicalId\":15888,\"journal\":{\"name\":\"Journal of Hepatology\",\"volume\":\"82 5\",\"pages\":\"Pages 871-881\"},\"PeriodicalIF\":33.0000,\"publicationDate\":\"2025-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Hepatology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0168827824027077\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"GASTROENTEROLOGY & HEPATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Hepatology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0168827824027077","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/15 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by inhibiting ATGL-mediated triglyceride hydrolysis
Background & Aims
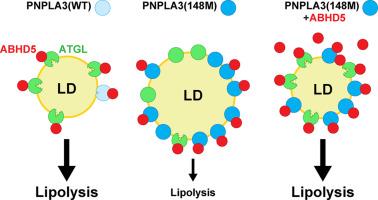
PNPLA3(148M) (patatin-like phospholipase domain-containing protein 3) is the most impactful genetic risk factor for steatotic liver disease. A key unresolved issue is whether PNPLA3(148M) confers a loss- or gain-of-function. Here we test the hypothesis that PNPLA3 causes steatosis by sequestering ABHD5 (α/β hydrolase domain-containing protein 5), the cofactor of ATGL (adipose TG lipase), thus limiting mobilization of hepatic triglyceride (TG).
Methods
We quantified and compared the physical interactions between ABHD5 and PNPLA3/ATGL in cultured hepatocytes using NanoBiT complementation assays and immunocytochemistry. Recombinant proteins purified from human cells were used to compare TG hydrolytic activities of PNPLA3 and ATGL in the presence or absence of ABHD5. Adenoviruses and adeno-associated viruses were used to express PNPLA3 in liver-specific Atgl-/- mice and to express ABHD5 in livers of Pnpla3M/M mice, respectively.
Results
ABHD5 interacted preferentially with PNPLA3 relative to ATGL in cultured hepatocytes. No differences were seen in the strength of the interactions between ABHD5 with PNPLA3(WT) and PNPLA3(148M). In contrast to prior findings, we found that PNPLA3, like ATGL, is activated by ABHD5 in in vitro assays using purified proteins. PNPLA3(148M)-associated inhibition of TG hydrolysis required that ATGL be expressed and that PNPLA3 be located on lipid droplets. Finally, overexpression of ABHD5 reversed the hepatic steatosis in Pnpla3M/M mice.
Conclusions
These findings support the premise that PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by accumulating on lipid droplets and inhibiting ATGL-mediated lipolysis in an ABHD5-dependent manner. Our results predict that reducing, rather than increasing, PNPLA3 expression will be the best strategy to treat PNPLA3(148M)-associated steatotic liver disease.
Impact and implications
Steatotic liver disease (SLD) is a common complex disorder associated with both environmental and genetic risk factors. PNPLA3(148M) is the most impactful genetic risk factor for SLD and yet its pathogenic mechanism remains controversial. Herein, we provide evidence that PNPLA3(148M) promotes triglyceride (TG) accumulation by sequestering ABHD5, thus limiting its availability to activate ATGL. Although the substitution of methionine for isoleucine reduces the TG hydrolase activity of PNPLA3, the loss of enzymatic function is not directly related to the steatotic effect of the variant. It is the resulting accumulation of PNPLA3 on LDs that confers a gain-of-function by interfering with ATGL-mediated TG hydrolysis. These findings have implications for the design of potential PNPLA3(148M)-based therapies. Reducing, rather than increasing, PNPLA3 levels is predicted to reverse steatosis in susceptible individuals.
期刊介绍:
The Journal of Hepatology is the official publication of the European Association for the Study of the Liver (EASL). It is dedicated to presenting clinical and basic research in the field of hepatology through original papers, reviews, case reports, and letters to the Editor. The Journal is published in English and may consider supplements that pass an editorial review.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
