{"title":"设计和研究卟啉席夫碱纳米片的电子状态,K+ - 电池的阴极材料","authors":"Fazal Dayan, Adnan Shahzad, Imad Ud Din","doi":"10.1016/j.comptc.2024.114936","DOIUrl":null,"url":null,"abstract":"<div><div>Identifying new electrode materials for K-ion batteries (KIBs) is still difficult since battery technology lacks an effective high-throughput screening approach. The durability, affordability, safety, and resemblance to Li-ion batteries of KIBs have garnered them tremendous attention. Porphyrin-based materials have become attractive options because of their generous surface area and advantageous photo-physical characteristics. As effective cathodic materials for KIBs, porphyrin Schiff base nanostructures (SBNs) are suggested in this work. Our goal was to improve Potassium-ion doped porphyrin derivatives by using Density Functional Theory (DFT) with the Gaussian 09 program with a B3LYP/6-31G(d) basis set. We also calculated important electronic parameters such as the band gap, electrophilicity, chemical potential, and frontier orbitals (HOMO and LUMO). The determined HOMO-LUMO gaps for compounds <strong>1</strong> to <strong>4</strong> were 2.97, 1.52, 1.38, and 1.36 respectively. The computed gaps indicate that the reactivity of the compounds increases from <strong>1</strong> to <strong>4</strong>, whereas the stability decreases from <strong>1</strong> to <strong>4</strong>.</div><div>Based on these computational observations, it is expected that this theoretical analysis will provide a basis for future researchers to explore the practical applications of these compounds through experimentation.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114936"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Designing and investigating electronic states of porphyrin Schiff bases nanoflakes, cathode materials for K+ - batteries\",\"authors\":\"Fazal Dayan, Adnan Shahzad, Imad Ud Din\",\"doi\":\"10.1016/j.comptc.2024.114936\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Identifying new electrode materials for K-ion batteries (KIBs) is still difficult since battery technology lacks an effective high-throughput screening approach. The durability, affordability, safety, and resemblance to Li-ion batteries of KIBs have garnered them tremendous attention. Porphyrin-based materials have become attractive options because of their generous surface area and advantageous photo-physical characteristics. As effective cathodic materials for KIBs, porphyrin Schiff base nanostructures (SBNs) are suggested in this work. Our goal was to improve Potassium-ion doped porphyrin derivatives by using Density Functional Theory (DFT) with the Gaussian 09 program with a B3LYP/6-31G(d) basis set. We also calculated important electronic parameters such as the band gap, electrophilicity, chemical potential, and frontier orbitals (HOMO and LUMO). The determined HOMO-LUMO gaps for compounds <strong>1</strong> to <strong>4</strong> were 2.97, 1.52, 1.38, and 1.36 respectively. The computed gaps indicate that the reactivity of the compounds increases from <strong>1</strong> to <strong>4</strong>, whereas the stability decreases from <strong>1</strong> to <strong>4</strong>.</div><div>Based on these computational observations, it is expected that this theoretical analysis will provide a basis for future researchers to explore the practical applications of these compounds through experimentation.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114936\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004754\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004754","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/7 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Designing and investigating electronic states of porphyrin Schiff bases nanoflakes, cathode materials for K+ - batteries
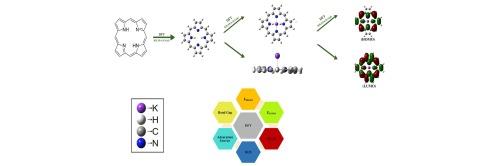
Identifying new electrode materials for K-ion batteries (KIBs) is still difficult since battery technology lacks an effective high-throughput screening approach. The durability, affordability, safety, and resemblance to Li-ion batteries of KIBs have garnered them tremendous attention. Porphyrin-based materials have become attractive options because of their generous surface area and advantageous photo-physical characteristics. As effective cathodic materials for KIBs, porphyrin Schiff base nanostructures (SBNs) are suggested in this work. Our goal was to improve Potassium-ion doped porphyrin derivatives by using Density Functional Theory (DFT) with the Gaussian 09 program with a B3LYP/6-31G(d) basis set. We also calculated important electronic parameters such as the band gap, electrophilicity, chemical potential, and frontier orbitals (HOMO and LUMO). The determined HOMO-LUMO gaps for compounds 1 to 4 were 2.97, 1.52, 1.38, and 1.36 respectively. The computed gaps indicate that the reactivity of the compounds increases from 1 to 4, whereas the stability decreases from 1 to 4.
Based on these computational observations, it is expected that this theoretical analysis will provide a basis for future researchers to explore the practical applications of these compounds through experimentation.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
