{"title":"复杂分子系统中电子激发的嵌入式多体格林函数方法","authors":"Gianluca Tirimbó, Vivek Sundaram, Björn Baumeier","doi":"10.1002/wcms.1734","DOIUrl":null,"url":null,"abstract":"<p>Many-body Green's function theory in the <i>GW</i> approximation with the Bethe–Salpeter equation (BSE) provides a powerful framework for the first-principles calculations of single-particle and electron–hole excitations in perfect crystals and molecules alike. Application to complex molecular systems, for example, solvated dyes, molecular aggregates, thin films, interfaces, or macromolecules, is particularly challenging as they contain a prohibitively large number of atoms. Exploiting the often localized nature of excitation in such disordered systems, several methods have recently been developed in which <i>GW</i>-BSE is applied to a smaller, tractable region of interest that is embedded into an environment described with a lower-level method. Here, we review the various strategies proposed for such embedded many-body Green's functions approaches, including quantum–quantum and quantum–classical embeddings, and focus in particular on how they include environment screening effects either intrinsically in the screened Coulomb interaction in the <i>GW</i> and BSE steps or via extrinsic electrostatic couplings.</p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 6","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2024-11-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1734","citationCount":"0","resultStr":"{\"title\":\"Embedded Many-Body Green's Function Methods for Electronic Excitations in Complex Molecular Systems\",\"authors\":\"Gianluca Tirimbó, Vivek Sundaram, Björn Baumeier\",\"doi\":\"10.1002/wcms.1734\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Many-body Green's function theory in the <i>GW</i> approximation with the Bethe–Salpeter equation (BSE) provides a powerful framework for the first-principles calculations of single-particle and electron–hole excitations in perfect crystals and molecules alike. Application to complex molecular systems, for example, solvated dyes, molecular aggregates, thin films, interfaces, or macromolecules, is particularly challenging as they contain a prohibitively large number of atoms. Exploiting the often localized nature of excitation in such disordered systems, several methods have recently been developed in which <i>GW</i>-BSE is applied to a smaller, tractable region of interest that is embedded into an environment described with a lower-level method. Here, we review the various strategies proposed for such embedded many-body Green's functions approaches, including quantum–quantum and quantum–classical embeddings, and focus in particular on how they include environment screening effects either intrinsically in the screened Coulomb interaction in the <i>GW</i> and BSE steps or via extrinsic electrostatic couplings.</p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 6\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2024-11-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1734\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1734\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1734","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Embedded Many-Body Green's Function Methods for Electronic Excitations in Complex Molecular Systems
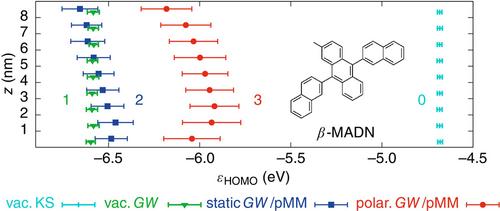
Many-body Green's function theory in the GW approximation with the Bethe–Salpeter equation (BSE) provides a powerful framework for the first-principles calculations of single-particle and electron–hole excitations in perfect crystals and molecules alike. Application to complex molecular systems, for example, solvated dyes, molecular aggregates, thin films, interfaces, or macromolecules, is particularly challenging as they contain a prohibitively large number of atoms. Exploiting the often localized nature of excitation in such disordered systems, several methods have recently been developed in which GW-BSE is applied to a smaller, tractable region of interest that is embedded into an environment described with a lower-level method. Here, we review the various strategies proposed for such embedded many-body Green's functions approaches, including quantum–quantum and quantum–classical embeddings, and focus in particular on how they include environment screening effects either intrinsically in the screened Coulomb interaction in the GW and BSE steps or via extrinsic electrostatic couplings.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
