Michael Iff, Kenneth Atz, Clemens Isert, Irene Pachon-Angona, Leandro Cotos, Mattis Hilleke, Jan A. Hiss and Gisbert Schneider
{"title":"将全新分子设计与半经验蛋白质配体结合自由能计算相结合†。","authors":"Michael Iff, Kenneth Atz, Clemens Isert, Irene Pachon-Angona, Leandro Cotos, Mattis Hilleke, Jan A. Hiss and Gisbert Schneider","doi":"10.1039/D4RA05422A","DOIUrl":null,"url":null,"abstract":"<p >Semi-empirical quantum chemistry methods estimate the binding free energies of protein–ligand complexes. We present an integrated approach combining the GFN2-<em>x</em>TB method with <em>de novo</em> design for the generation and evaluation of potential inhibitors of acetylcholinesterase (AChE). We employed chemical language model-based molecule generation to explore the synthetically accessible chemical space around the natural product Huperzine A, a potent AChE inhibitor. Four distinct molecular libraries were created using structure- and ligand-based molecular <em>de novo</em> design with SMILES and SELFIES representations, respectively. These libraries were computationally evaluated for synthesizability, novelty, and predicted biological activity. The candidate molecules were subjected to molecular docking to identify hypothetical binding poses, which were further refined using Gibbs free energy calculations. The structurally novel top-ranked molecule was chemically synthesized and biologically tested, demonstrating moderate micromolar activity against AChE. Our findings highlight the potential and certain limitations of integrating deep learning-based molecular generation with semi-empirical quantum chemistry-based activity prediction for structure-based drug design.</p>","PeriodicalId":102,"journal":{"name":"RSC Advances","volume":" 50","pages":" 37035-37044"},"PeriodicalIF":6.1000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/ra/d4ra05422a?page=search","citationCount":"0","resultStr":"{\"title\":\"Combining de novo molecular design with semiempirical protein–ligand binding free energy calculation†\",\"authors\":\"Michael Iff, Kenneth Atz, Clemens Isert, Irene Pachon-Angona, Leandro Cotos, Mattis Hilleke, Jan A. Hiss and Gisbert Schneider\",\"doi\":\"10.1039/D4RA05422A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Semi-empirical quantum chemistry methods estimate the binding free energies of protein–ligand complexes. We present an integrated approach combining the GFN2-<em>x</em>TB method with <em>de novo</em> design for the generation and evaluation of potential inhibitors of acetylcholinesterase (AChE). We employed chemical language model-based molecule generation to explore the synthetically accessible chemical space around the natural product Huperzine A, a potent AChE inhibitor. Four distinct molecular libraries were created using structure- and ligand-based molecular <em>de novo</em> design with SMILES and SELFIES representations, respectively. These libraries were computationally evaluated for synthesizability, novelty, and predicted biological activity. The candidate molecules were subjected to molecular docking to identify hypothetical binding poses, which were further refined using Gibbs free energy calculations. The structurally novel top-ranked molecule was chemically synthesized and biologically tested, demonstrating moderate micromolar activity against AChE. Our findings highlight the potential and certain limitations of integrating deep learning-based molecular generation with semi-empirical quantum chemistry-based activity prediction for structure-based drug design.</p>\",\"PeriodicalId\":102,\"journal\":{\"name\":\"RSC Advances\",\"volume\":\" 50\",\"pages\":\" 37035-37044\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2024-11-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/ra/d4ra05422a?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"RSC Advances\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/ra/d4ra05422a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Advances","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ra/d4ra05422a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Combining de novo molecular design with semiempirical protein–ligand binding free energy calculation†
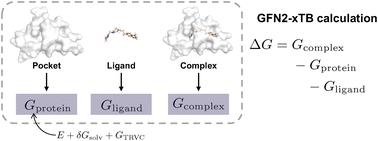
Semi-empirical quantum chemistry methods estimate the binding free energies of protein–ligand complexes. We present an integrated approach combining the GFN2-xTB method with de novo design for the generation and evaluation of potential inhibitors of acetylcholinesterase (AChE). We employed chemical language model-based molecule generation to explore the synthetically accessible chemical space around the natural product Huperzine A, a potent AChE inhibitor. Four distinct molecular libraries were created using structure- and ligand-based molecular de novo design with SMILES and SELFIES representations, respectively. These libraries were computationally evaluated for synthesizability, novelty, and predicted biological activity. The candidate molecules were subjected to molecular docking to identify hypothetical binding poses, which were further refined using Gibbs free energy calculations. The structurally novel top-ranked molecule was chemically synthesized and biologically tested, demonstrating moderate micromolar activity against AChE. Our findings highlight the potential and certain limitations of integrating deep learning-based molecular generation with semi-empirical quantum chemistry-based activity prediction for structure-based drug design.
期刊介绍:
An international, peer-reviewed journal covering all of the chemical sciences, including multidisciplinary and emerging areas. RSC Advances is a gold open access journal allowing researchers free access to research articles, and offering an affordable open access publishing option for authors around the world.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
