José Leonardo Gómez Chávez, Adriano Martín Luchi, Roxana Noelia Villafañe, Germán Andres Conti, Ernesto Rafael Perez, Emilio Luis Angelina, Nélida María Peruchena
{"title":"图神经网络和分子对接是虚拟筛选的两种互补方法:克鲁扎因的案例研究","authors":"José Leonardo Gómez Chávez, Adriano Martín Luchi, Roxana Noelia Villafañe, Germán Andres Conti, Ernesto Rafael Perez, Emilio Luis Angelina, Nélida María Peruchena","doi":"10.1002/slct.202405342","DOIUrl":null,"url":null,"abstract":"<p>Molecular docking is one of the most widely used techniques for virtual screening (VS) of potential drug candidates. Despite its popularity, docking accuracy is often limited due to the trade-off between speed and precision required for screening large compound libraries. In the present work, we leverage graph convolutional networks (GCNs), a state-of-the-art deep neural network architecture, to enhance docking capacity for prioritizing active compounds from a library of ∼200,000 compounds screened against Cruzain. We propose strategies to integrate both techniques into a single VS pipeline. By applying the GCN as a pre-docking filter, the compound library was enriched with active molecules, resulting in higher hit rates in subsequent docking screenings. Additionally, to further enhance the docking performance, the GCN-learned atomic embeddings were directly incorporated into the docking process through pharmacophoric restraints. Unlike common approaches that use deep learning (DL) scoring functions to rank pre-generated docking poses, the approaches we propose here have the advantage that only compounds that passed the DL filters need to be screened by the more computationally demanding docking method. This work might serve as a proof of concept for combining deep learning and classical docking in drug discovery.</p>","PeriodicalId":146,"journal":{"name":"ChemistrySelect","volume":"9 44","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-11-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Graph Neural Networks and Molecular Docking as Two Complementary Approaches for Virtual Screening: A Case Study on Cruzain\",\"authors\":\"José Leonardo Gómez Chávez, Adriano Martín Luchi, Roxana Noelia Villafañe, Germán Andres Conti, Ernesto Rafael Perez, Emilio Luis Angelina, Nélida María Peruchena\",\"doi\":\"10.1002/slct.202405342\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Molecular docking is one of the most widely used techniques for virtual screening (VS) of potential drug candidates. Despite its popularity, docking accuracy is often limited due to the trade-off between speed and precision required for screening large compound libraries. In the present work, we leverage graph convolutional networks (GCNs), a state-of-the-art deep neural network architecture, to enhance docking capacity for prioritizing active compounds from a library of ∼200,000 compounds screened against Cruzain. We propose strategies to integrate both techniques into a single VS pipeline. By applying the GCN as a pre-docking filter, the compound library was enriched with active molecules, resulting in higher hit rates in subsequent docking screenings. Additionally, to further enhance the docking performance, the GCN-learned atomic embeddings were directly incorporated into the docking process through pharmacophoric restraints. Unlike common approaches that use deep learning (DL) scoring functions to rank pre-generated docking poses, the approaches we propose here have the advantage that only compounds that passed the DL filters need to be screened by the more computationally demanding docking method. This work might serve as a proof of concept for combining deep learning and classical docking in drug discovery.</p>\",\"PeriodicalId\":146,\"journal\":{\"name\":\"ChemistrySelect\",\"volume\":\"9 44\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-11-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ChemistrySelect\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202405342\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemistrySelect","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202405342","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
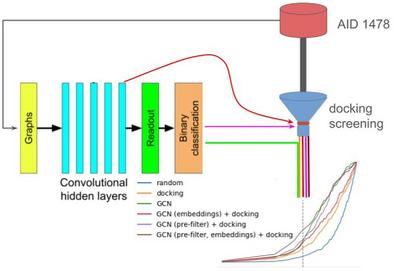
分子对接是对潜在候选药物进行虚拟筛选(VS)的最广泛应用技术之一。尽管它很受欢迎,但由于筛选大型化合物库所需的速度和精度之间的权衡,对接的准确性往往受到限制。在本研究中,我们利用最先进的深度神经网络架构--图卷积网络(GCNs)来提高对接能力,以便从针对克鲁赛因筛选的 20 万个化合物库中优先筛选出活性化合物。我们提出了将这两种技术整合到单一 VS 管线中的策略。通过应用 GCN 作为预对接过滤器,化合物库中的活性分子更加丰富,从而提高了后续对接筛选的命中率。此外,为了进一步提高对接性能,GCN 学习到的原子嵌入通过药理学约束直接纳入对接过程。与使用深度学习(DL)评分函数对预先生成的对接姿势进行排序的常见方法不同,我们在此提出的方法的优势在于,只有通过 DL 筛选的化合物才需要通过计算要求更高的对接方法进行筛选。这项工作可以作为在药物发现中结合深度学习和经典对接的概念验证。
Graph Neural Networks and Molecular Docking as Two Complementary Approaches for Virtual Screening: A Case Study on Cruzain
Molecular docking is one of the most widely used techniques for virtual screening (VS) of potential drug candidates. Despite its popularity, docking accuracy is often limited due to the trade-off between speed and precision required for screening large compound libraries. In the present work, we leverage graph convolutional networks (GCNs), a state-of-the-art deep neural network architecture, to enhance docking capacity for prioritizing active compounds from a library of ∼200,000 compounds screened against Cruzain. We propose strategies to integrate both techniques into a single VS pipeline. By applying the GCN as a pre-docking filter, the compound library was enriched with active molecules, resulting in higher hit rates in subsequent docking screenings. Additionally, to further enhance the docking performance, the GCN-learned atomic embeddings were directly incorporated into the docking process through pharmacophoric restraints. Unlike common approaches that use deep learning (DL) scoring functions to rank pre-generated docking poses, the approaches we propose here have the advantage that only compounds that passed the DL filters need to be screened by the more computationally demanding docking method. This work might serve as a proof of concept for combining deep learning and classical docking in drug discovery.
期刊介绍:
ChemistrySelect is the latest journal from ChemPubSoc Europe and Wiley-VCH. It offers researchers a quality society-owned journal in which to publish their work in all areas of chemistry. Manuscripts are evaluated by active researchers to ensure they add meaningfully to the scientific literature, and those accepted are processed quickly to ensure rapid online publication.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
