Christopher P. Earl, Mathias Cobbaut, André Barros-Carvalho, Marina E. Ivanova, David C. Briggs, Eurico Morais-de-Sá, Peter J. Parker, Neil Q. McDonald
{"title":"aPKC-Par6及其多位点极性底物Lgl的捕获、相互抑制和释放机制","authors":"Christopher P. Earl, Mathias Cobbaut, André Barros-Carvalho, Marina E. Ivanova, David C. Briggs, Eurico Morais-de-Sá, Peter J. Parker, Neil Q. McDonald","doi":"10.1038/s41594-024-01425-0","DOIUrl":null,"url":null,"abstract":"The mutually antagonistic relationship of atypical protein kinase C (aPKC) and partitioning-defective protein 6 (Par6) with the substrate lethal (2) giant larvae (Lgl) is essential for regulating polarity across many cell types. Although aPKC–Par6 phosphorylates Lgl at three serine sites to exclude it from the apical domain, aPKC–Par6 and Lgl paradoxically form a stable kinase–substrate complex, with conflicting roles proposed for Par6. We report the structure of human aPKCι–Par6α bound to full-length Llgl1, captured through an aPKCι docking site and a Par6PDZ contact. This complex traps a phospho-S663 Llgl1 intermediate bridging between aPKC and Par6, impeding phosphorylation progression. Thus, aPKCι is effectively inhibited by Llgl1pS663 while Llgl1 is captured by aPKCι–Par6. Mutational disruption of the Lgl–aPKC interaction impedes complex assembly and Lgl phosphorylation, whereas disrupting the Lgl–Par6PDZ contact promotes complex dissociation and Lgl phosphorylation. We demonstrate a Par6PDZ-regulated substrate capture-and-release model requiring binding by active Cdc42 and the apical partner Crumbs to drive complex disassembly. Our results suggest a mechanism for mutual regulation and spatial control of aPKC–Par6 and Lgl activities. Combining structural, biochemical, cellular and in vivo assays, the authors uncover the mechanism for capture and multisite phosphorylation of lethal (2) giant larvae by the atypical protein kinase C and partitioning-defective protein 6, revealing the basis for their mutual antagonism underpinning cell polarity.","PeriodicalId":49141,"journal":{"name":"Nature Structural & Molecular Biology","volume":"32 4","pages":"729-739"},"PeriodicalIF":10.1000,"publicationDate":"2025-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.comhttps://www.nature.com/articles/s41594-024-01425-0.pdf","citationCount":"0","resultStr":"{\"title\":\"Capture, mutual inhibition and release mechanism for aPKC–Par6 and its multisite polarity substrate Lgl\",\"authors\":\"Christopher P. Earl, Mathias Cobbaut, André Barros-Carvalho, Marina E. Ivanova, David C. Briggs, Eurico Morais-de-Sá, Peter J. Parker, Neil Q. McDonald\",\"doi\":\"10.1038/s41594-024-01425-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The mutually antagonistic relationship of atypical protein kinase C (aPKC) and partitioning-defective protein 6 (Par6) with the substrate lethal (2) giant larvae (Lgl) is essential for regulating polarity across many cell types. Although aPKC–Par6 phosphorylates Lgl at three serine sites to exclude it from the apical domain, aPKC–Par6 and Lgl paradoxically form a stable kinase–substrate complex, with conflicting roles proposed for Par6. We report the structure of human aPKCι–Par6α bound to full-length Llgl1, captured through an aPKCι docking site and a Par6PDZ contact. This complex traps a phospho-S663 Llgl1 intermediate bridging between aPKC and Par6, impeding phosphorylation progression. Thus, aPKCι is effectively inhibited by Llgl1pS663 while Llgl1 is captured by aPKCι–Par6. Mutational disruption of the Lgl–aPKC interaction impedes complex assembly and Lgl phosphorylation, whereas disrupting the Lgl–Par6PDZ contact promotes complex dissociation and Lgl phosphorylation. We demonstrate a Par6PDZ-regulated substrate capture-and-release model requiring binding by active Cdc42 and the apical partner Crumbs to drive complex disassembly. Our results suggest a mechanism for mutual regulation and spatial control of aPKC–Par6 and Lgl activities. Combining structural, biochemical, cellular and in vivo assays, the authors uncover the mechanism for capture and multisite phosphorylation of lethal (2) giant larvae by the atypical protein kinase C and partitioning-defective protein 6, revealing the basis for their mutual antagonism underpinning cell polarity.\",\"PeriodicalId\":49141,\"journal\":{\"name\":\"Nature Structural & Molecular Biology\",\"volume\":\"32 4\",\"pages\":\"729-739\"},\"PeriodicalIF\":10.1000,\"publicationDate\":\"2025-01-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.comhttps://www.nature.com/articles/s41594-024-01425-0.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Structural & Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41594-024-01425-0\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Structural & Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41594-024-01425-0","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Capture, mutual inhibition and release mechanism for aPKC–Par6 and its multisite polarity substrate Lgl
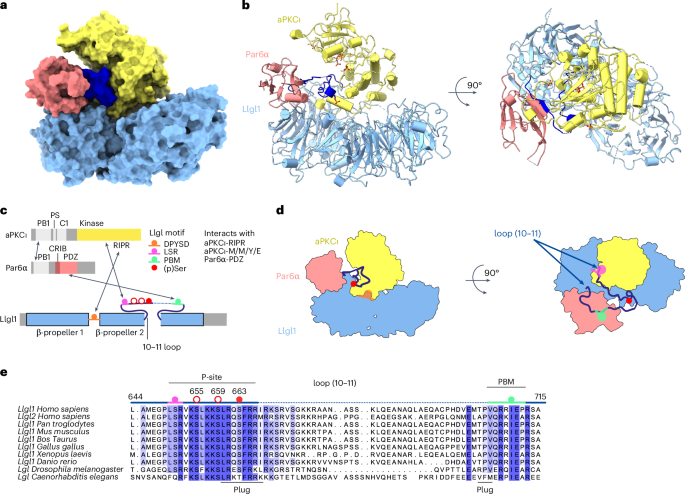
The mutually antagonistic relationship of atypical protein kinase C (aPKC) and partitioning-defective protein 6 (Par6) with the substrate lethal (2) giant larvae (Lgl) is essential for regulating polarity across many cell types. Although aPKC–Par6 phosphorylates Lgl at three serine sites to exclude it from the apical domain, aPKC–Par6 and Lgl paradoxically form a stable kinase–substrate complex, with conflicting roles proposed for Par6. We report the structure of human aPKCι–Par6α bound to full-length Llgl1, captured through an aPKCι docking site and a Par6PDZ contact. This complex traps a phospho-S663 Llgl1 intermediate bridging between aPKC and Par6, impeding phosphorylation progression. Thus, aPKCι is effectively inhibited by Llgl1pS663 while Llgl1 is captured by aPKCι–Par6. Mutational disruption of the Lgl–aPKC interaction impedes complex assembly and Lgl phosphorylation, whereas disrupting the Lgl–Par6PDZ contact promotes complex dissociation and Lgl phosphorylation. We demonstrate a Par6PDZ-regulated substrate capture-and-release model requiring binding by active Cdc42 and the apical partner Crumbs to drive complex disassembly. Our results suggest a mechanism for mutual regulation and spatial control of aPKC–Par6 and Lgl activities. Combining structural, biochemical, cellular and in vivo assays, the authors uncover the mechanism for capture and multisite phosphorylation of lethal (2) giant larvae by the atypical protein kinase C and partitioning-defective protein 6, revealing the basis for their mutual antagonism underpinning cell polarity.
期刊介绍:
Nature Structural & Molecular Biology is a comprehensive platform that combines structural and molecular research. Our journal focuses on exploring the functional and mechanistic aspects of biological processes, emphasizing how molecular components collaborate to achieve a particular function. While structural data can shed light on these insights, our publication does not require them as a prerequisite.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
