Cheryl Shoubridge, May Huey Tan, Tod Fullston, Desiree Cloosterman, David Coman, George McGillivray, Grazia M Mancini, Tjitske Kleefstra, Jozef Gécz
{"title":"Aristaless相关同型盒核定位序列突变含有IPO13的ARX突变体的隔离破坏了转录因子的正常亚细胞分布并延缓了细胞分裂。","authors":"Cheryl Shoubridge, May Huey Tan, Tod Fullston, Desiree Cloosterman, David Coman, George McGillivray, Grazia M Mancini, Tjitske Kleefstra, Jozef Gécz","doi":"10.1186/1755-8417-3-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Aristaless related homeobox (ARX) is a paired-type homeobox gene. ARX function is frequently affected by naturally occurring mutations. Nonsense mutations, polyalanine tract expansions and missense mutations in ARX cause a range of intellectual disability and epilepsy phenotypes with or without additional features including hand dystonia, lissencephaly, autism or dysarthria. Severe malformation phenotypes, such as X-linked lissencephaly with ambiguous genitalia (XLAG), are frequently observed in individuals with protein truncating or missense mutations clustered in the highly conserved paired-type homeodomain.</p><p><strong>Results: </strong>We have identified two novel point mutations in the R379 residue of the ARX homeodomain; c.1135C>A, p.R379S in a patient with infantile spasms and intellectual disability and c.1136G>T, p.R379L in a patient with XLAG. We investigated these and other missense mutations (R332P, R332H, R332C, T333N: associated with XLAG and Proud syndrome) predicted to affect the nuclear localisation sequences (NLS) flanking either end of the ARX homeodomain. The NLS regions are required for correct nuclear import facilitated by Importin 13 (IPO13). We demonstrate that missense mutations in either the N- or C-terminal NLS regions of the homeodomain cause significant disruption to nuclear localisation of the ARX protein in vitro. Surprisingly, none of these mutations abolished the binding of ARX to IPO13. This was confirmed by co-immunoprecipitation and immmuno fluorescence studies. Instead, tagged and endogenous IPO13 remained bound to the mutant ARX proteins, even in the RanGTP rich nuclear environment. We also identify the microtubule protein TUBA1A as a novel interacting protein for ARX and show cells expressing mutant ARX protein accumulate in mitosis, indicating normal cell division may be disrupted.</p><p><strong>Conclusions: </strong>We show that the most likely, common pathogenic mechanism of the missense mutations in NLS regions of the ARX homeodomain is inadequate accumulation and distribution of the ARX transcription factor within the nucleus due to sequestration of ARX with IPO13.</p>","PeriodicalId":88084,"journal":{"name":"PathoGenetics","volume":"3 ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2010-01-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1755-8417-3-1","citationCount":"37","resultStr":"{\"title\":\"Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division.\",\"authors\":\"Cheryl Shoubridge, May Huey Tan, Tod Fullston, Desiree Cloosterman, David Coman, George McGillivray, Grazia M Mancini, Tjitske Kleefstra, Jozef Gécz\",\"doi\":\"10.1186/1755-8417-3-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Aristaless related homeobox (ARX) is a paired-type homeobox gene. ARX function is frequently affected by naturally occurring mutations. Nonsense mutations, polyalanine tract expansions and missense mutations in ARX cause a range of intellectual disability and epilepsy phenotypes with or without additional features including hand dystonia, lissencephaly, autism or dysarthria. Severe malformation phenotypes, such as X-linked lissencephaly with ambiguous genitalia (XLAG), are frequently observed in individuals with protein truncating or missense mutations clustered in the highly conserved paired-type homeodomain.</p><p><strong>Results: </strong>We have identified two novel point mutations in the R379 residue of the ARX homeodomain; c.1135C>A, p.R379S in a patient with infantile spasms and intellectual disability and c.1136G>T, p.R379L in a patient with XLAG. We investigated these and other missense mutations (R332P, R332H, R332C, T333N: associated with XLAG and Proud syndrome) predicted to affect the nuclear localisation sequences (NLS) flanking either end of the ARX homeodomain. The NLS regions are required for correct nuclear import facilitated by Importin 13 (IPO13). We demonstrate that missense mutations in either the N- or C-terminal NLS regions of the homeodomain cause significant disruption to nuclear localisation of the ARX protein in vitro. Surprisingly, none of these mutations abolished the binding of ARX to IPO13. This was confirmed by co-immunoprecipitation and immmuno fluorescence studies. Instead, tagged and endogenous IPO13 remained bound to the mutant ARX proteins, even in the RanGTP rich nuclear environment. We also identify the microtubule protein TUBA1A as a novel interacting protein for ARX and show cells expressing mutant ARX protein accumulate in mitosis, indicating normal cell division may be disrupted.</p><p><strong>Conclusions: </strong>We show that the most likely, common pathogenic mechanism of the missense mutations in NLS regions of the ARX homeodomain is inadequate accumulation and distribution of the ARX transcription factor within the nucleus due to sequestration of ARX with IPO13.</p>\",\"PeriodicalId\":88084,\"journal\":{\"name\":\"PathoGenetics\",\"volume\":\"3 \",\"pages\":\"1\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2010-01-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1755-8417-3-1\",\"citationCount\":\"37\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PathoGenetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1755-8417-3-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PathoGenetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1755-8417-3-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 37
摘要
背景:ARX (aristoess related homeobox)是一种配对型同源盒基因。ARX功能经常受到自然发生的突变的影响。ARX的无义突变、多丙氨酸束扩张和错义突变导致一系列智力残疾和癫痫表型,伴有或不伴有其他特征,包括手肌张力障碍、无脑畸形、自闭症或构音障碍。严重的畸形表型,如带有模糊生殖器的x连锁无脑畸形(XLAG),经常在高度保守的成对型同源结构域中聚集的蛋白质截断或错义突变的个体中观察到。结果:我们在ARX同源域的R379残基上发现了两个新的点突变;c.1135C>A, p.R379S在婴儿痉挛和智力残疾患者中,c.1136G>T, p.R379L在XLAG患者中。我们研究了这些和其他误义突变(R332P, R332H, R332C, T333N:与XLAG和Proud综合征相关)预测会影响ARX同源结构域两侧的核定位序列(NLS)。NLS地区需要通过importin13 (IPO13)促进正确的核进口。在体外实验中,我们证明了同位结构域N端或c端NLS区域的错义突变会对ARX蛋白的核定位造成显著的破坏。令人惊讶的是,这些突变都没有破坏ARX与IPO13的结合。免疫共沉淀和免疫荧光研究证实了这一点。相反,即使在富含RanGTP的核环境中,标记的内源性IPO13仍然与突变的ARX蛋白结合。我们还发现微管蛋白TUBA1A是一种新的ARX相互作用蛋白,并显示表达突变ARX蛋白的细胞在有丝分裂中积累,表明正常的细胞分裂可能被破坏。结论:我们发现,ARX同位结构域NLS区错义突变最可能的常见致病机制是由于IPO13对ARX的隔离导致ARX转录因子在细胞核内的积累和分布不足。
Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division.
Background: Aristaless related homeobox (ARX) is a paired-type homeobox gene. ARX function is frequently affected by naturally occurring mutations. Nonsense mutations, polyalanine tract expansions and missense mutations in ARX cause a range of intellectual disability and epilepsy phenotypes with or without additional features including hand dystonia, lissencephaly, autism or dysarthria. Severe malformation phenotypes, such as X-linked lissencephaly with ambiguous genitalia (XLAG), are frequently observed in individuals with protein truncating or missense mutations clustered in the highly conserved paired-type homeodomain.
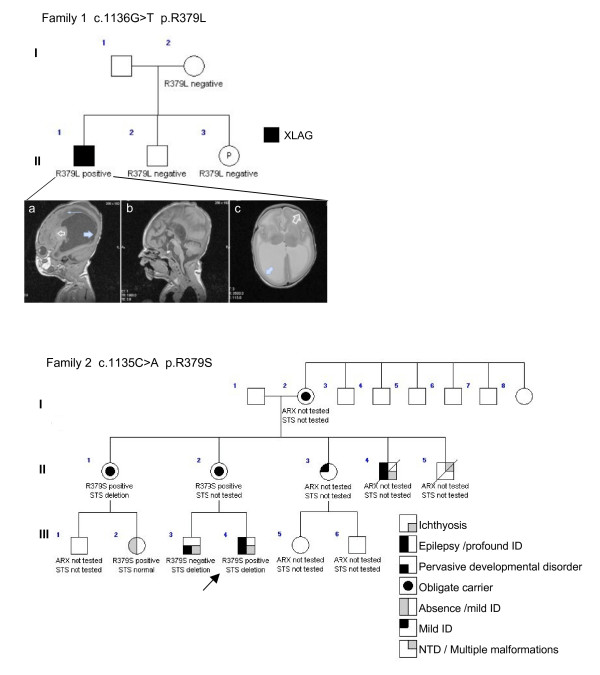
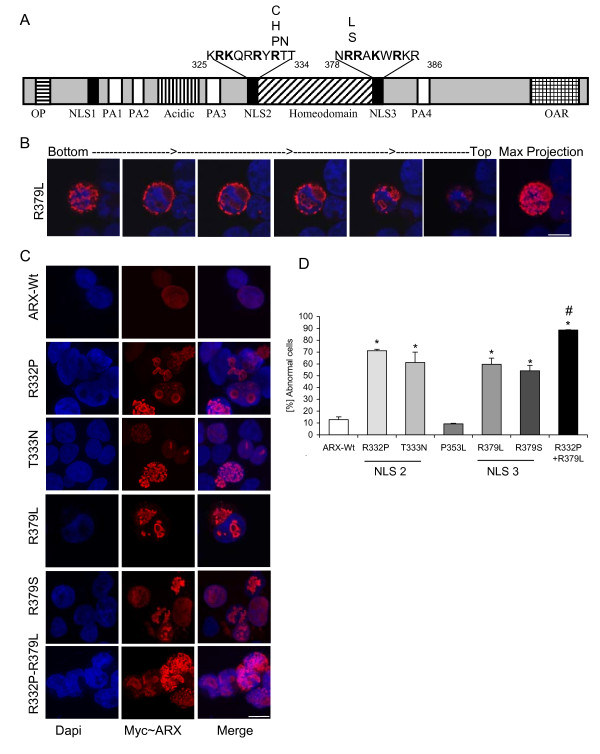
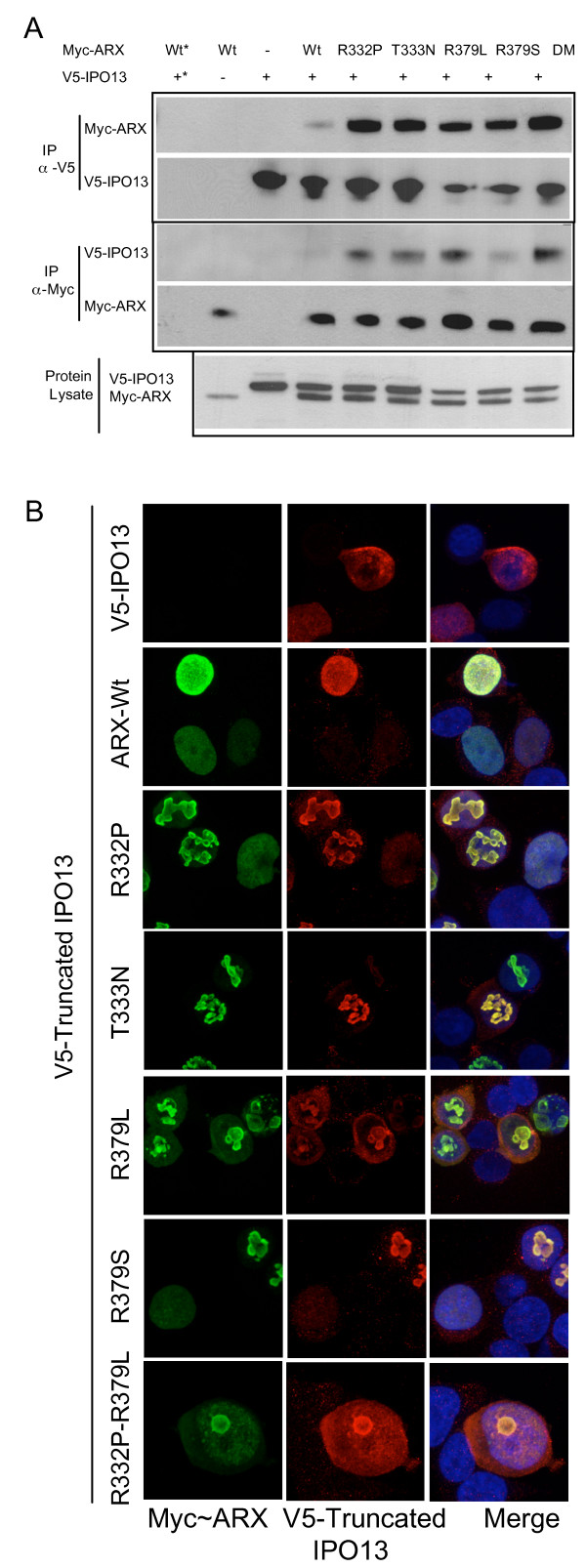
Results: We have identified two novel point mutations in the R379 residue of the ARX homeodomain; c.1135C>A, p.R379S in a patient with infantile spasms and intellectual disability and c.1136G>T, p.R379L in a patient with XLAG. We investigated these and other missense mutations (R332P, R332H, R332C, T333N: associated with XLAG and Proud syndrome) predicted to affect the nuclear localisation sequences (NLS) flanking either end of the ARX homeodomain. The NLS regions are required for correct nuclear import facilitated by Importin 13 (IPO13). We demonstrate that missense mutations in either the N- or C-terminal NLS regions of the homeodomain cause significant disruption to nuclear localisation of the ARX protein in vitro. Surprisingly, none of these mutations abolished the binding of ARX to IPO13. This was confirmed by co-immunoprecipitation and immmuno fluorescence studies. Instead, tagged and endogenous IPO13 remained bound to the mutant ARX proteins, even in the RanGTP rich nuclear environment. We also identify the microtubule protein TUBA1A as a novel interacting protein for ARX and show cells expressing mutant ARX protein accumulate in mitosis, indicating normal cell division may be disrupted.
Conclusions: We show that the most likely, common pathogenic mechanism of the missense mutations in NLS regions of the ARX homeodomain is inadequate accumulation and distribution of the ARX transcription factor within the nucleus due to sequestration of ARX with IPO13.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
