{"title":"使用稀疏方法揭示神经胶质瘤异质性的基于转录组学的特征。","authors":"Sofia Martins, Roberta Coletti, Marta B Lopes","doi":"10.1186/s13040-023-00341-1","DOIUrl":null,"url":null,"abstract":"<p><p>Gliomas are primary malignant brain tumors with poor survival and high resistance to available treatments. Improving the molecular understanding of glioma and disclosing novel biomarkers of tumor development and progression could help to find novel targeted therapies for this type of cancer. Public databases such as The Cancer Genome Atlas (TCGA) provide an invaluable source of molecular information on cancer tissues. Machine learning tools show promise in dealing with the high dimension of omics data and extracting relevant information from it. In this work, network inference and clustering methods, namely Joint Graphical lasso and Robust Sparse K-means Clustering, were applied to RNA-sequencing data from TCGA glioma patients to identify shared and distinct gene networks among different types of glioma (glioblastoma, astrocytoma, and oligodendroglioma) and disclose new patient groups and the relevant genes behind groups' separation. The results obtained suggest that astrocytoma and oligodendroglioma have more similarities compared with glioblastoma, highlighting the molecular differences between glioblastoma and the others glioma subtypes. After a comprehensive literature search on the relevant genes pointed our from our analysis, we identified potential candidates for biomarkers of glioma. Further molecular validation of these genes is encouraged to understand their potential role in diagnosis and in the design of novel therapies.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"26"},"PeriodicalIF":6.1000,"publicationDate":"2023-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10523751/pdf/","citationCount":"0","resultStr":"{\"title\":\"Disclosing transcriptomics network-based signatures of glioma heterogeneity using sparse methods.\",\"authors\":\"Sofia Martins, Roberta Coletti, Marta B Lopes\",\"doi\":\"10.1186/s13040-023-00341-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Gliomas are primary malignant brain tumors with poor survival and high resistance to available treatments. Improving the molecular understanding of glioma and disclosing novel biomarkers of tumor development and progression could help to find novel targeted therapies for this type of cancer. Public databases such as The Cancer Genome Atlas (TCGA) provide an invaluable source of molecular information on cancer tissues. Machine learning tools show promise in dealing with the high dimension of omics data and extracting relevant information from it. In this work, network inference and clustering methods, namely Joint Graphical lasso and Robust Sparse K-means Clustering, were applied to RNA-sequencing data from TCGA glioma patients to identify shared and distinct gene networks among different types of glioma (glioblastoma, astrocytoma, and oligodendroglioma) and disclose new patient groups and the relevant genes behind groups' separation. The results obtained suggest that astrocytoma and oligodendroglioma have more similarities compared with glioblastoma, highlighting the molecular differences between glioblastoma and the others glioma subtypes. After a comprehensive literature search on the relevant genes pointed our from our analysis, we identified potential candidates for biomarkers of glioma. Further molecular validation of these genes is encouraged to understand their potential role in diagnosis and in the design of novel therapies.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\"16 1\",\"pages\":\"26\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2023-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10523751/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-023-00341-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00341-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Disclosing transcriptomics network-based signatures of glioma heterogeneity using sparse methods.
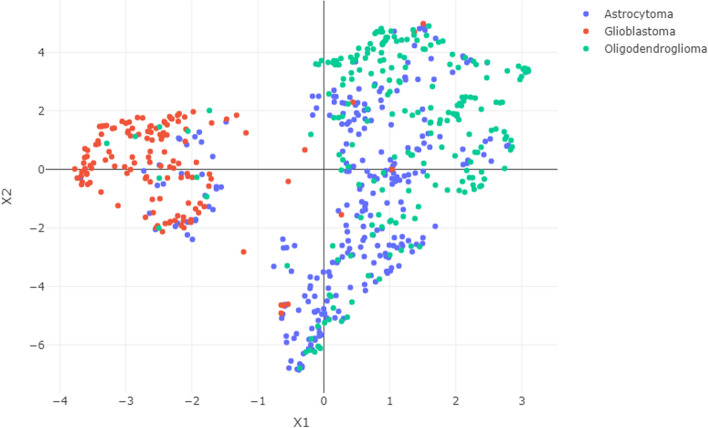
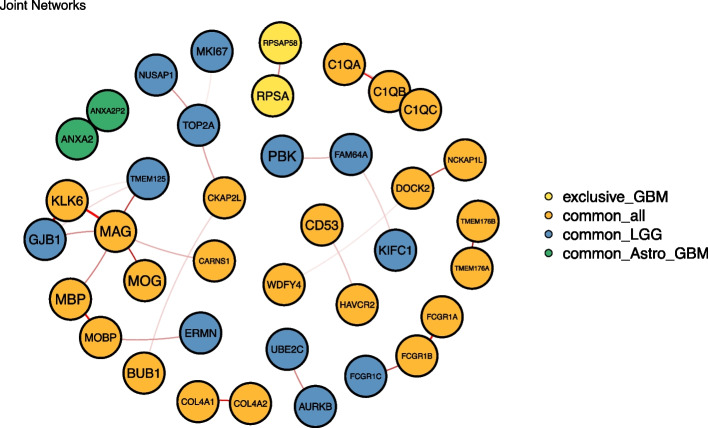
Gliomas are primary malignant brain tumors with poor survival and high resistance to available treatments. Improving the molecular understanding of glioma and disclosing novel biomarkers of tumor development and progression could help to find novel targeted therapies for this type of cancer. Public databases such as The Cancer Genome Atlas (TCGA) provide an invaluable source of molecular information on cancer tissues. Machine learning tools show promise in dealing with the high dimension of omics data and extracting relevant information from it. In this work, network inference and clustering methods, namely Joint Graphical lasso and Robust Sparse K-means Clustering, were applied to RNA-sequencing data from TCGA glioma patients to identify shared and distinct gene networks among different types of glioma (glioblastoma, astrocytoma, and oligodendroglioma) and disclose new patient groups and the relevant genes behind groups' separation. The results obtained suggest that astrocytoma and oligodendroglioma have more similarities compared with glioblastoma, highlighting the molecular differences between glioblastoma and the others glioma subtypes. After a comprehensive literature search on the relevant genes pointed our from our analysis, we identified potential candidates for biomarkers of glioma. Further molecular validation of these genes is encouraged to understand their potential role in diagnosis and in the design of novel therapies.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
