{"title":"MM/GBSA预测碳酸酐酶抑制剂的相对结合亲和力:原子电荷的影响以及与Autodock4Zn的比较","authors":"Mackenzie Taylor, Junming Ho","doi":"10.1007/s10822-023-00499-0","DOIUrl":null,"url":null,"abstract":"<div><p>Carbonic anhydrase is an attractive drug target for the treatment of many diseases. This paper examines the ability of end-state MM/GBSA methods to rank inhibitors of carbonic anhydrase in terms of their binding affinities. The MM/GBSA binding energies were evaluated using different atomic charge schemes (Mulliken, ESP and NPA) at different levels of theories, including Hartree–Fock, B3LYP-D3(BJ), and M06-2X with the 6–31G(d,p) basis set. For a large test set of 32 diverse inhibitors, the use of B3LYP-D3(BJ) ESP atomic charges yielded the strongest correlation with experiment (R<sup>2</sup> = 0.77). The use of the recently enhanced Autodock Vina and zinc optimised AD4<sub>Zn</sub> force field also predicted ligand binding affinities with moderately strong correlation (R<sup>2</sup> = 0.64) at significantly lower computational cost. However, the docked poses deviate significantly from crystal structures. Overall, this study demonstrates the applicability of docking to estimate ligand binding affinities for a diverse range of CA inhibitors, and indicates that more theoretically robust MM/GBSA simulations show promise for improving the accuracy of predicted binding affinities, as long as a validated set of parameters is used.</p><h3>Graphical abstract</h3>\n <figure><div><div><div><picture><source><img></source></picture></div></div></div></figure>\n </div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 4","pages":"167 - 182"},"PeriodicalIF":3.1000,"publicationDate":"2023-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00499-0.pdf","citationCount":"0","resultStr":"{\"title\":\"MM/GBSA prediction of relative binding affinities of carbonic anhydrase inhibitors: effect of atomic charges and comparison with Autodock4Zn\",\"authors\":\"Mackenzie Taylor, Junming Ho\",\"doi\":\"10.1007/s10822-023-00499-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Carbonic anhydrase is an attractive drug target for the treatment of many diseases. This paper examines the ability of end-state MM/GBSA methods to rank inhibitors of carbonic anhydrase in terms of their binding affinities. The MM/GBSA binding energies were evaluated using different atomic charge schemes (Mulliken, ESP and NPA) at different levels of theories, including Hartree–Fock, B3LYP-D3(BJ), and M06-2X with the 6–31G(d,p) basis set. For a large test set of 32 diverse inhibitors, the use of B3LYP-D3(BJ) ESP atomic charges yielded the strongest correlation with experiment (R<sup>2</sup> = 0.77). The use of the recently enhanced Autodock Vina and zinc optimised AD4<sub>Zn</sub> force field also predicted ligand binding affinities with moderately strong correlation (R<sup>2</sup> = 0.64) at significantly lower computational cost. However, the docked poses deviate significantly from crystal structures. Overall, this study demonstrates the applicability of docking to estimate ligand binding affinities for a diverse range of CA inhibitors, and indicates that more theoretically robust MM/GBSA simulations show promise for improving the accuracy of predicted binding affinities, as long as a validated set of parameters is used.</p><h3>Graphical abstract</h3>\\n <figure><div><div><div><picture><source><img></source></picture></div></div></div></figure>\\n </div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 4\",\"pages\":\"167 - 182\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-03-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-023-00499-0.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00499-0\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00499-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
MM/GBSA prediction of relative binding affinities of carbonic anhydrase inhibitors: effect of atomic charges and comparison with Autodock4Zn
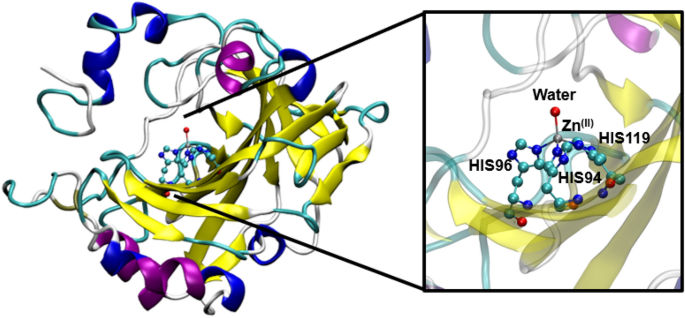
Carbonic anhydrase is an attractive drug target for the treatment of many diseases. This paper examines the ability of end-state MM/GBSA methods to rank inhibitors of carbonic anhydrase in terms of their binding affinities. The MM/GBSA binding energies were evaluated using different atomic charge schemes (Mulliken, ESP and NPA) at different levels of theories, including Hartree–Fock, B3LYP-D3(BJ), and M06-2X with the 6–31G(d,p) basis set. For a large test set of 32 diverse inhibitors, the use of B3LYP-D3(BJ) ESP atomic charges yielded the strongest correlation with experiment (R2 = 0.77). The use of the recently enhanced Autodock Vina and zinc optimised AD4Zn force field also predicted ligand binding affinities with moderately strong correlation (R2 = 0.64) at significantly lower computational cost. However, the docked poses deviate significantly from crystal structures. Overall, this study demonstrates the applicability of docking to estimate ligand binding affinities for a diverse range of CA inhibitors, and indicates that more theoretically robust MM/GBSA simulations show promise for improving the accuracy of predicted binding affinities, as long as a validated set of parameters is used.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
